

The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes
- PMID: 18436705
- PMCID: PMC2528859
- DOI: 10.1210/er.2007-0039
The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes
Erratum in
- Endocr Rev. 2008 Aug;29(5):631
Abstract
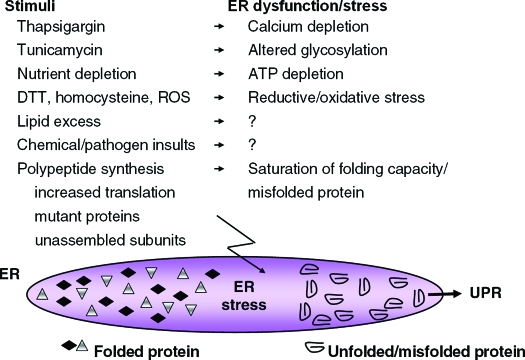
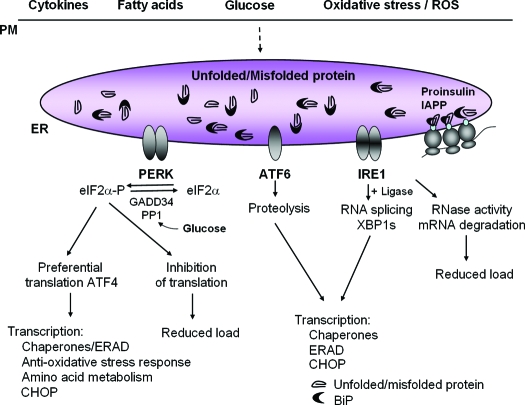
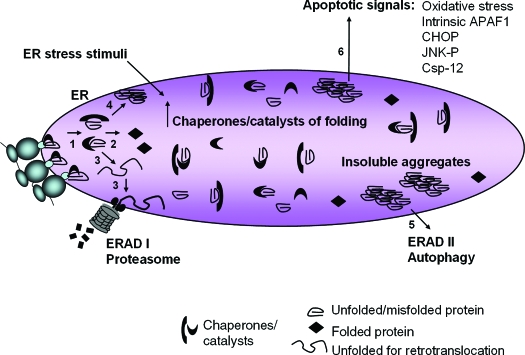
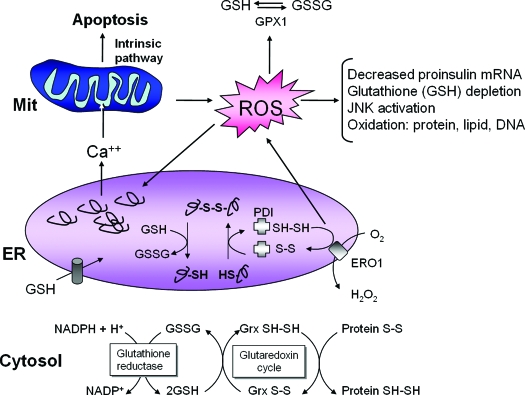
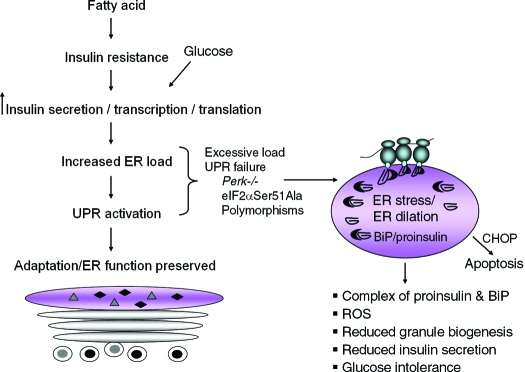
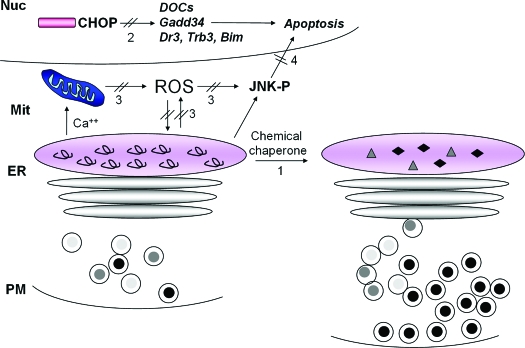
The endoplasmic reticulum (ER) is the entry site into the secretory pathway for newly synthesized proteins destined for the cell surface or released into the extracellular milieu. The study of protein folding and trafficking within the ER is an extremely active area of research that has provided novel insights into many disease processes. Cells have evolved mechanisms to modulate the capacity and quality of the ER protein-folding machinery to prevent the accumulation of unfolded or misfolded proteins. These signaling pathways are collectively termed the unfolded protein response (UPR). The UPR sensors signal a transcriptional response to expand the ER folding capacity, increase degradation of malfolded proteins, and limit the rate of mRNA translation to reduce the client protein load. Recent genetic and biochemical evidence in both humans and mice supports a requirement for the UPR to preserve ER homeostasis and prevent the beta-cell failure that may be fundamental in the etiology of diabetes. Chronic or overwhelming ER stress stimuli associated with metabolic syndrome can disrupt protein folding in the ER, reduce insulin secretion, invoke oxidative stress, and activate cell death pathways. Therapeutic interventions to prevent polypeptide-misfolding, oxidative damage, and/or UPR-induced cell death have the potential to improve beta-cell function and/or survival in the treatment of diabetes.
Figures
References
-
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS 2003 Global analysis of protein expression in yeast. Nature 425:737–741 - PubMed
-
- Ma Y, Hendershot LM 2004 ER chaperone functions during normal and stress conditions. J Chem Neuroanat 28:51–65 - PubMed
-
- Kleizen B, Braakman I 2004 Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol 16:343–349 - PubMed
-
- Ellgaard L 2004 Catalysis of disulphide bond formation in the endoplasmic reticulum. Biochem Soc Trans 32:663–667 - PubMed
-
- Sevier CS, Kaiser CA 2006 Conservation and diversity of the cellular disulfide bond formation pathways. Antioxid Redox Signal 8:797–811 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
