

Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy
- PMID: 18440926
- PMCID: PMC4908855
- DOI: 10.1126/science.1155085
Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy
Erratum in
-
Erratum for the Report "Plastin 3 Is a Protective Modifier of Autosomal Recessive Spinal Muscular Atrophy" by G. E. Oprea et al.Science. 2024 May 31;384(6699):eadq4773. doi: 10.1126/science.adq4773. Epub 2024 May 30. Science. 2024. PMID: 38815046 No abstract available.
Abstract
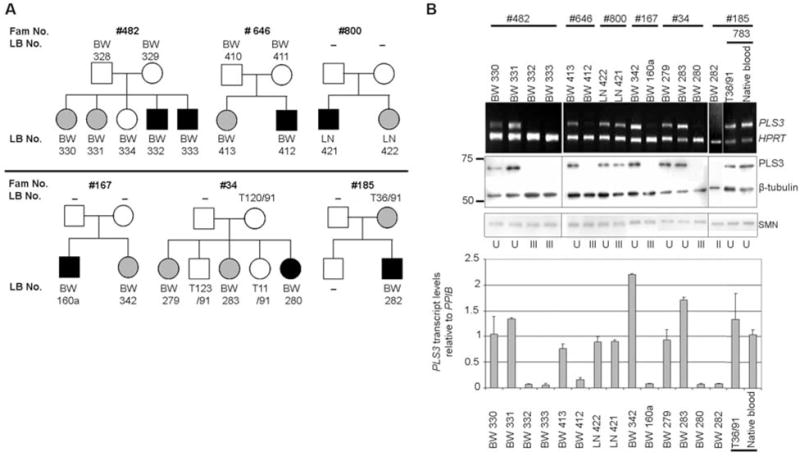
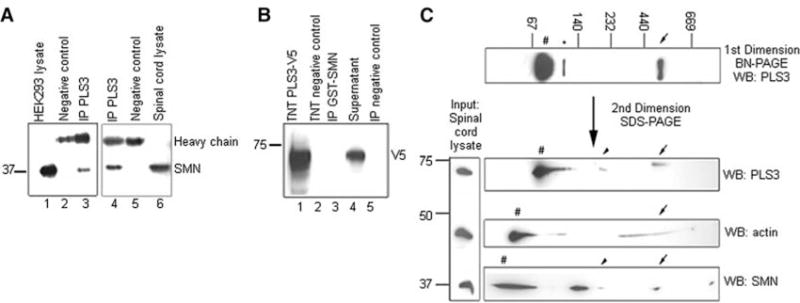
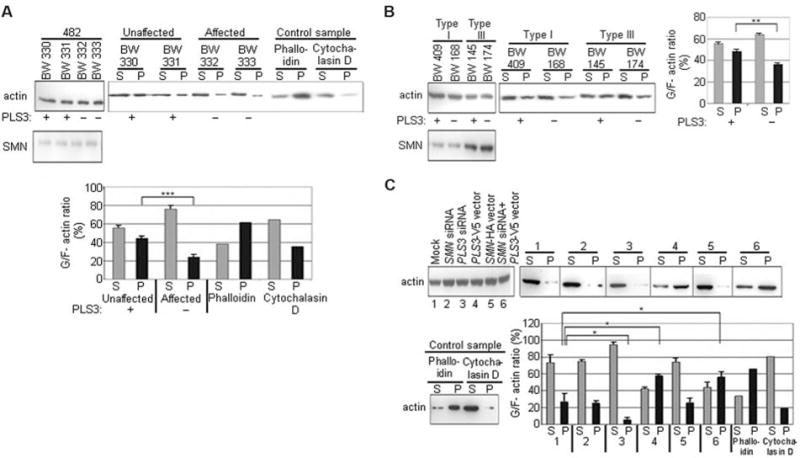
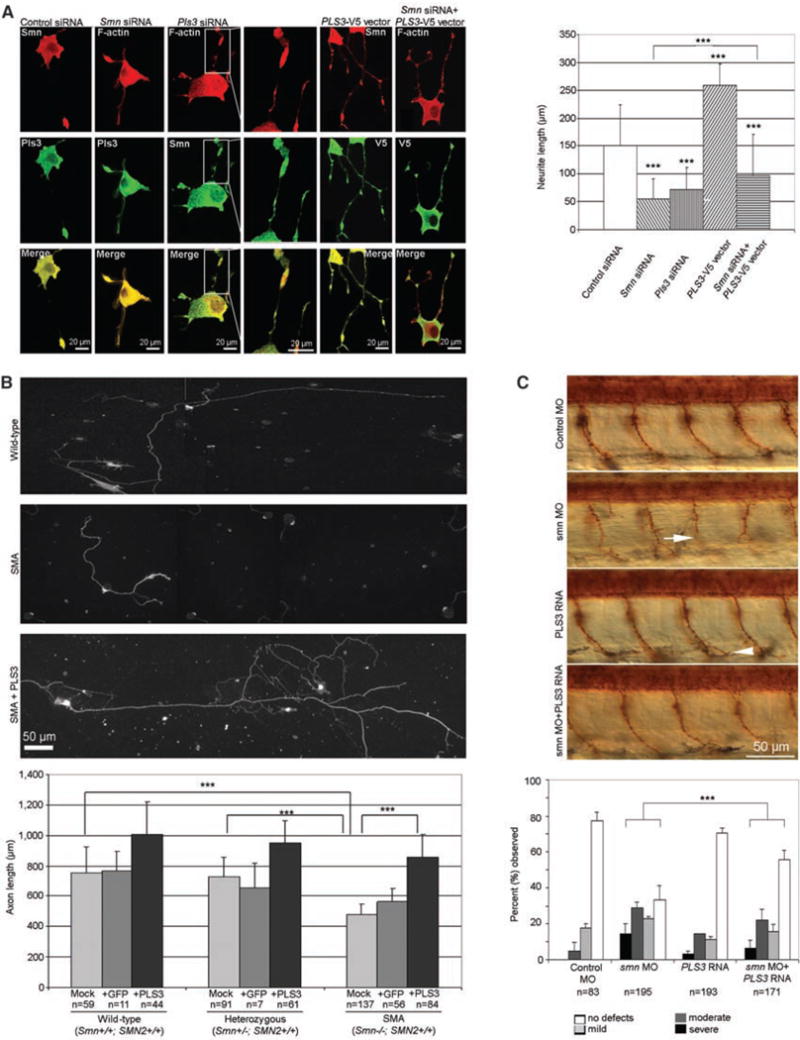
Homozygous deletion of the survival motor neuron 1 gene (SMN1) causes spinal muscular atrophy (SMA), the most frequent genetic cause of early childhood lethality. In rare instances, however, individuals are asymptomatic despite carrying the same SMN1 mutations as their affected siblings, thereby suggesting the influence of modifier genes. We discovered that unaffected SMN1-deleted females exhibit significantly higher expression of plastin 3 (PLS3) than their SMA-affected counterparts. We demonstrated that PLS3 is important for axonogenesis through increasing the F-actin level. Overexpression of PLS3 rescued the axon length and outgrowth defects associated with SMN down-regulation in motor neurons of SMA mouse embryos and in zebrafish. Our study suggests that defects in axonogenesis are the major cause of SMA, thereby opening new therapeutic options for SMA and similar neuromuscular diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
