

Acute effects of oestrogen on the guinea pig and human IKr channels and drug-induced prolongation of cardiac repolarization
- PMID: 18440994
- PMCID: PMC2517194
- DOI: 10.1113/jphysiol.2007.150367
Acute effects of oestrogen on the guinea pig and human IKr channels and drug-induced prolongation of cardiac repolarization
Abstract
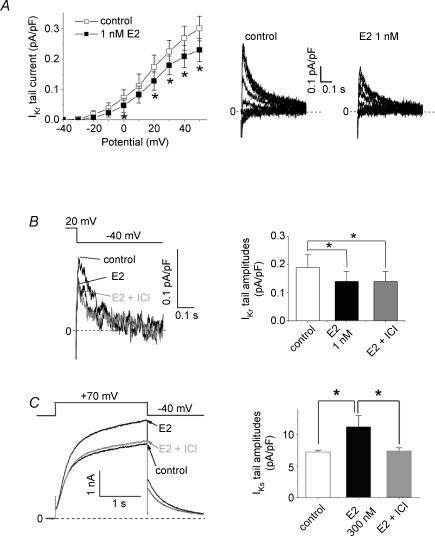
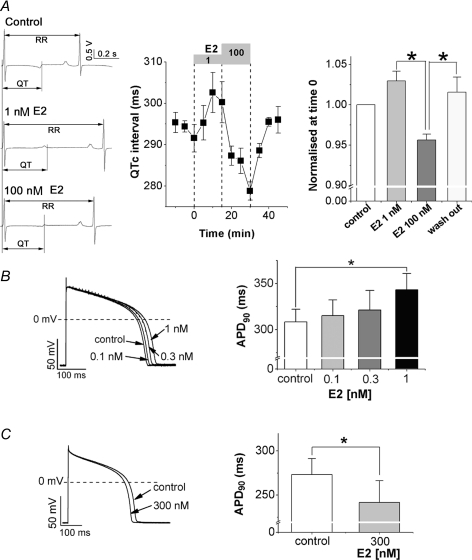
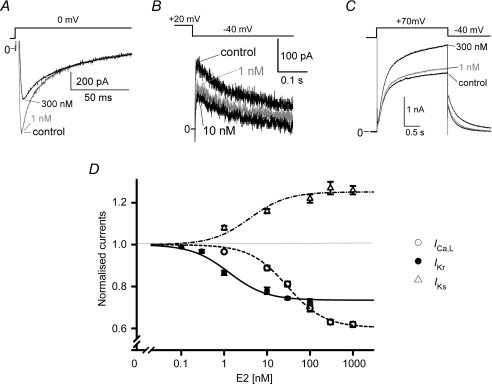
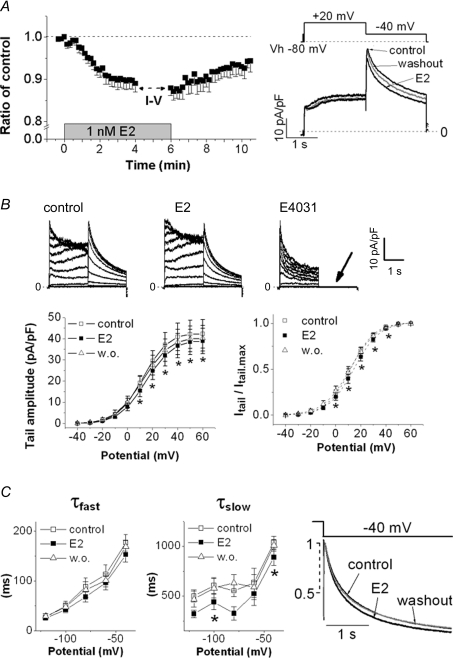
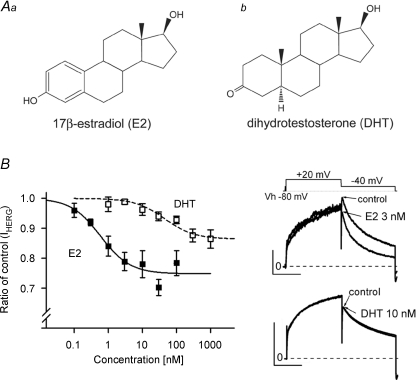
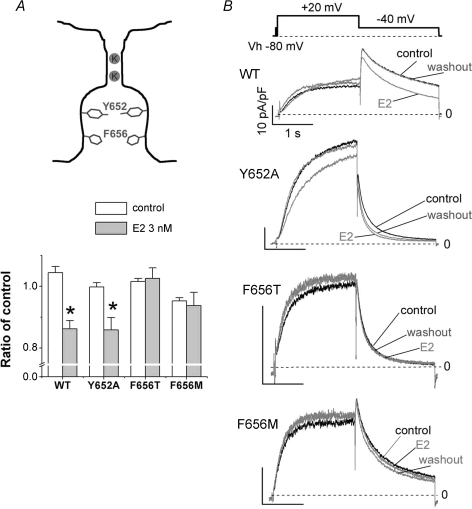
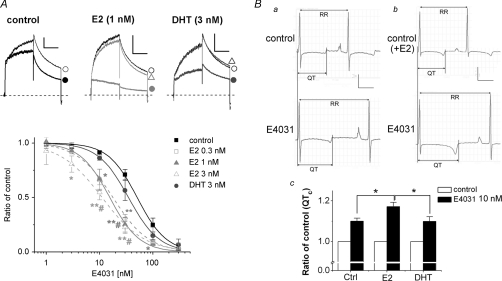
Female gender is a risk factor for drug-induced arrhythmias associated with QT prolongation, which results mostly from blockade of the human ether-a-go-go-related gene (hERG) channel. Some clinical evidence suggests that oestrogen is a determinant of the gender-differences in drug-induced QT prolongation and baseline QT(C) intervals. Although the chronic effects of oestrogen have been studied, it remains unclear whether the gender differences are due entirely to transcriptional regulations through oestrogen receptors. We therefore investigated acute effects of the most bioactive oestrogen, 17beta-oestradiol (E2) at its physiological concentrations on cardiac repolarization and drug-sensitivity of the hERG (I(Kr)) channel in Langendorff-perfused guinea pig hearts, patch-clamped guinea pig cardiomyocytes and culture cells over-expressing hERG. We found that physiological concentrations of E2 partially suppressed I(Kr) in a receptor-independent manner. E2-induced modification of voltage-dependence causes partial suppression of hERG currents. Mutagenesis studies showed that a common drug-binding residue at the inner pore cavity was critical for the effects of E2 on the hERG channel. Furthermore, E2 enhanced both hERG suppression and QT(C) prolongation by its blocker, E4031. The lack of effects of testosterone at its physiological concentrations on both of hERG currents and E4031-sensitivity of the hERG channel implicates the critical role of aromatic centroid present in E2 but not in testosterone. Our data indicate that E2 acutely affects the hERG channel gating and the E4031-induced QT(C) prolongation, and may provide a novel mechanism for the higher susceptibility to drug-induced arrhythmia in women.
Figures
Comment in
-
Male and female equality: still far from goal.J Physiol. 2008 Jun 15;586(12):2825-6. doi: 10.1113/jphysiol.2008.156398. J Physiol. 2008. PMID: 18556724 Free PMC article. No abstract available.
References
-
- Abi-Gerges N, Philp K, Pollard C, Wakefield I, Hammond TG, Valentin JP. Sex differences in ventricular repolarization: from cardiac electrophysiology to Torsades de Pointes. Fundam Clin Pharmacol. 2004;18:139–151. - PubMed
-
- Bai CX, Kurokawa J, Tamagawa M, Nakaya H, Furukawa T. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation. 2005;112:1701–1710. - PubMed
-
- Clancy CE, Kurokawa J, Tateyama M, Wehrens XH, Kass RS. K+ channel structure-activity relationships and mechanisms of drug-induced QT prolongation. Annu Rev Pharmacol Toxicol. 2003;43:441–461. - PubMed
-
- Drici MD, Burklow TR, Haridasse V, Glazer RI, Woosley RL. Sex hormones prolong the QT interval and downregulate potassium channel expression in the rabbit heart. Circulation. 1996;94:1471–1474. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources