

Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation
- PMID: 18441292
- PMCID: PMC2364605
- DOI: 10.1101/lm.984508
Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation
Abstract
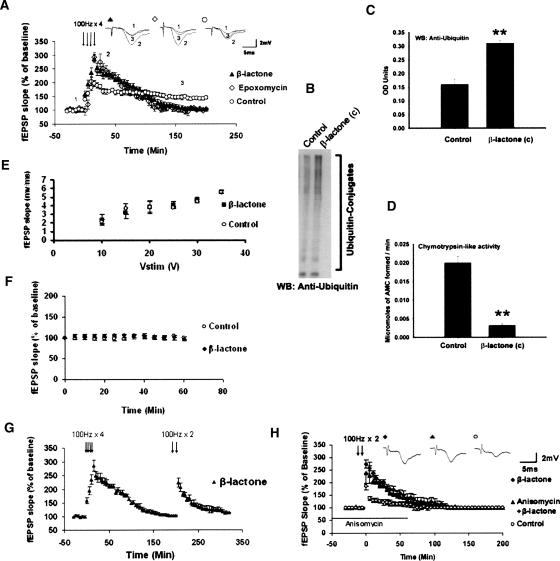
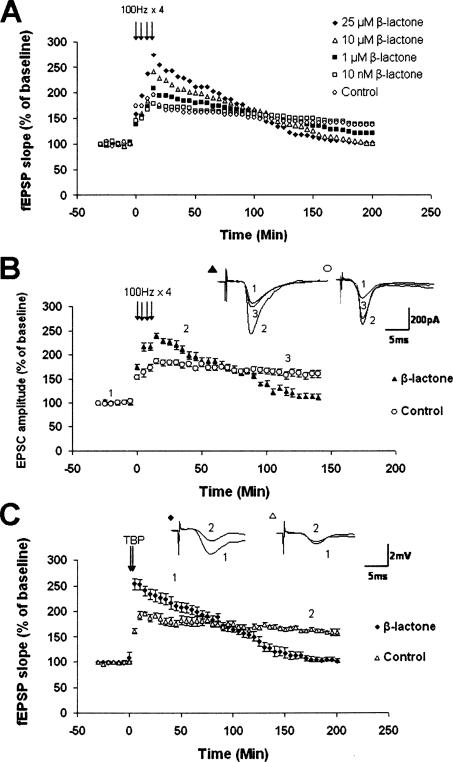
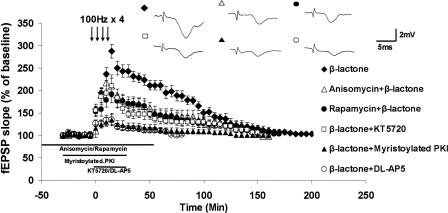
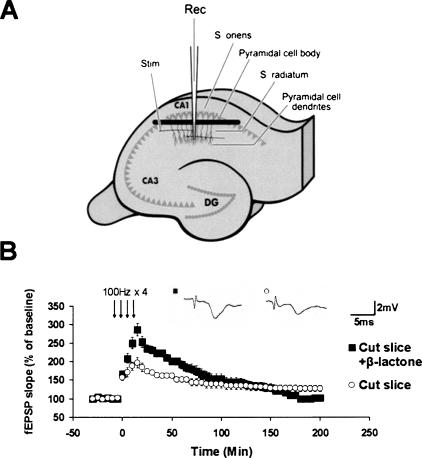
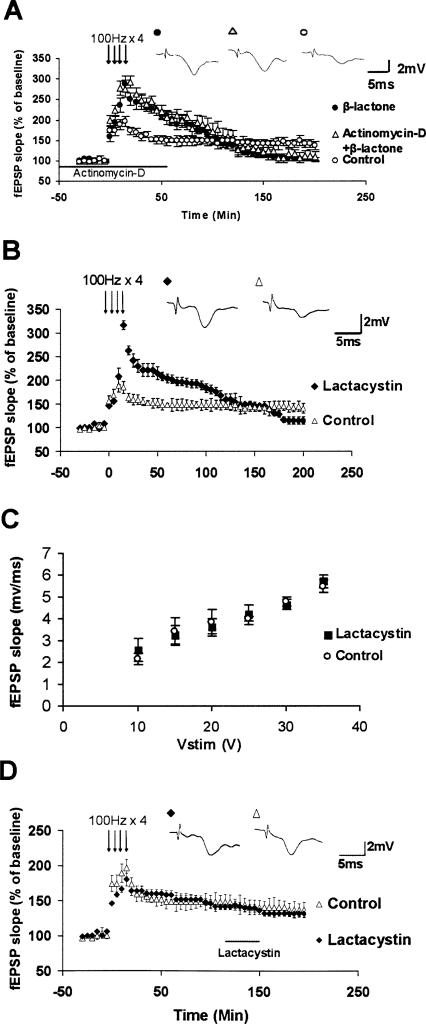
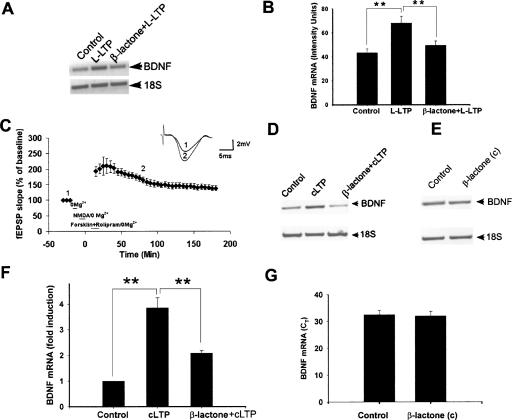
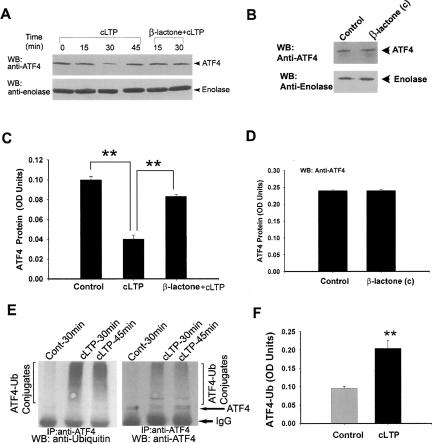
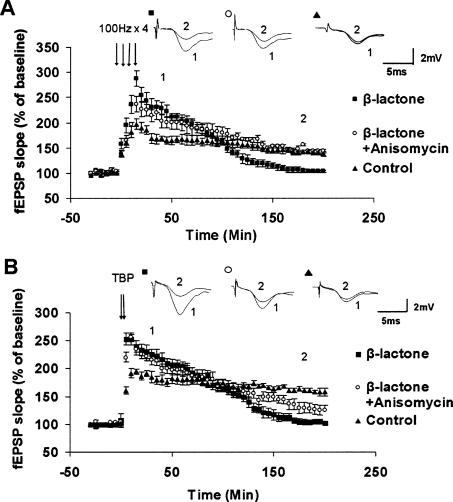
Protein degradation by the ubiquitin-proteasome pathway plays important roles in synaptic plasticity, but the molecular mechanisms by which proteolysis regulates synaptic strength are not well understood. We investigated the role of the proteasome in hippocampal late-phase long-term potentiation (L-LTP), a model for enduring synaptic plasticity. We show here that inhibition of the proteasome enhances the induction of L-LTP, but inhibits its maintenance. Proteasome inhibitor-mediated enhancement of the early part of L-LTP requires activation of NMDA receptors and the cAMP-dependent protein kinase. Augmentation of L-LTP induction by proteasome inhibition is blocked by a protein synthesis inhibitor anisomycin and is sensitive to the drug rapamycin. Our findings indicate that proteasome inhibition increases the induction of L-LTP by stabilizing locally translated proteins in dendrites. In addition, our data show that inhibition of the proteasome blocks transcription of brain-derived neurotrophic factor (BDNF), which is a cAMP-responsive element-binding protein (CREB)-inducible gene. Furthermore, our results demonstrate that the proteasome inhibitors block degradation of ATF4, a CREB repressor. Thus, proteasome inhibition appears to hinder CREB-mediated transcription. Our results indicate that blockade of proteasome activity obstructs the maintenance of L-LTP by interfering with transcription as well as translation required to sustain L-LTP. Thus, proteasome-mediated proteolysis has different roles during the induction and the maintenance of L-LTP.
Figures
References
-
- Abel T., Nguyen P.V., Barad M., Deuel T.A., Kandel E.R., Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. - PubMed
-
- Barco A., Alarcon J.M., Kandel E.R. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. - PubMed
-
- Barco A., Patterson S., Alarcon J.M., Gromova P., Mata-Roig M., Morozov A., Kandel E.R. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 2005;48:123–137. - PubMed
-
- Bland B.H. The physiology and pharmacology of hippocampal formation theta rhythms. Prog. Neurobiol. 1986;26:1–54. - PubMed
-
- Blitzer R.D., Iyengar R., Landau E.M. Postsynaptic signaling networks: Cellular cogwheels underlying long-term plasticity. Biol. Psychiatry. 2005;57:113–119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources