

Direct binding of integrin alphavbeta3 to FGF1 plays a role in FGF1 signaling
- PMID: 18441324
- PMCID: PMC2440593
- DOI: 10.1074/jbc.M801213200
Direct binding of integrin alphavbeta3 to FGF1 plays a role in FGF1 signaling
Abstract
Integrins play a role in fibroblast growth factor (FGF) signaling through cross-talk with FGF receptors (FGFRs), but the mechanism underlying the cross-talk is unknown. We discovered that FGF1 directly bound to soluble and cell-surface integrin alphavbeta3 (K(D) about 1 microm). Antagonists to alphavbeta3 (monoclonal antibody 7E3 and cyclic RGDfV) blocked this interaction. alphavbeta3 was the predominant, if not the only, integrin that bound to FGF1, because FGF1 bound only weakly to several beta1 integrins tested. We presented evidence that the CYDMKTTC sequence (the specificity loop) within the ligand-binding site of beta3 plays a role in FGF1 binding. We found that the integrin-binding site of FGF1 overlaps with the heparin-binding site but is distinct from the FGFR-binding site using docking simulation and mutagenesis. We identified an FGF1 mutant (R50E) that was defective in integrin binding but still bound to heparin and FGFR. R50E was defective in inducing DNA synthesis, cell proliferation, cell migration, and chemotaxis, suggesting that the direct integrin binding to FGF1 is critical for FGF signaling. Nevertheless, R50E induced phosphorylation of FGFR1 and FRS2alpha and activation of AKT and ERK1/2. These results suggest that the defect in R50E in FGF signaling is not in the initial activation of FGF signaling pathway components, but in the later steps in FGF signaling. We propose that R50E is a useful tool to identify the role of integrins in FGF signaling.
Figures
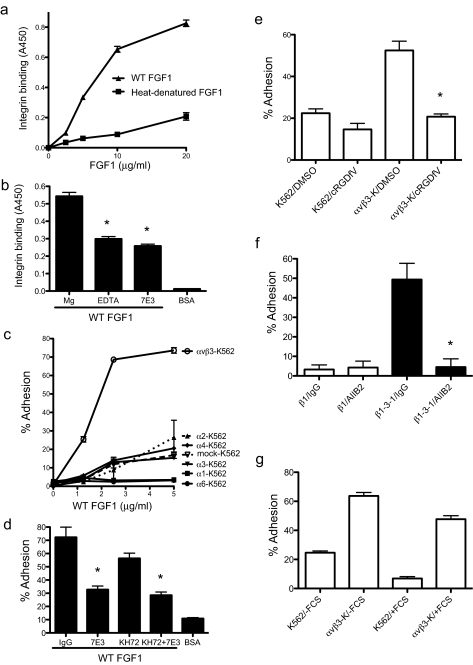
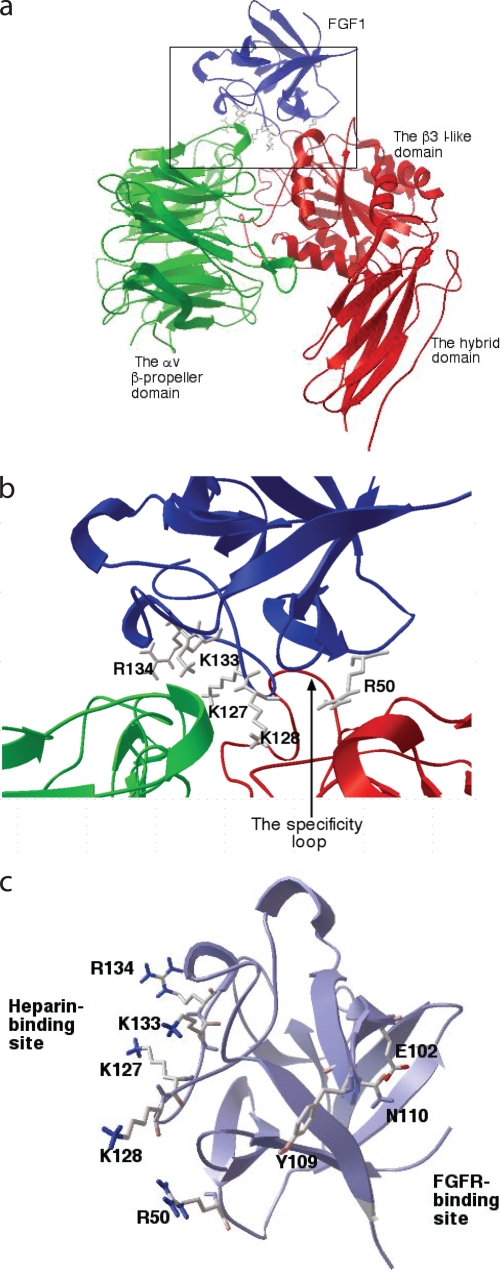
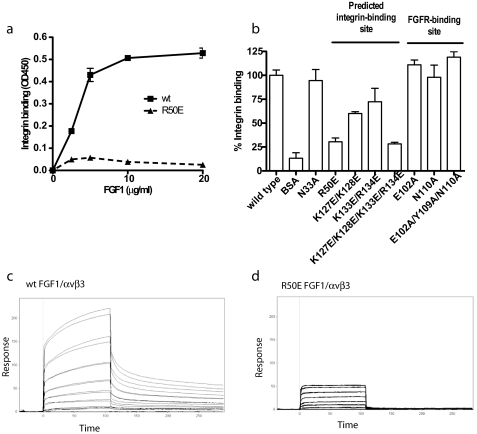
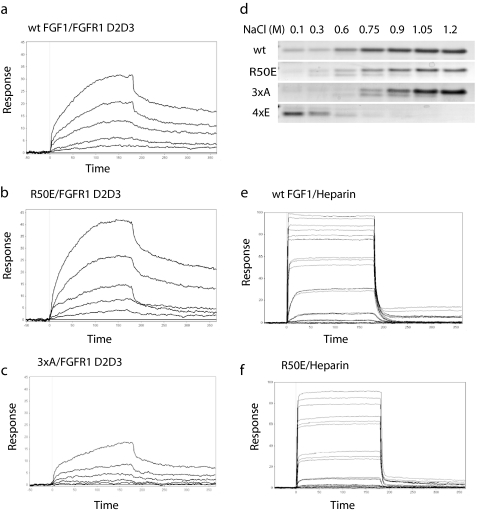
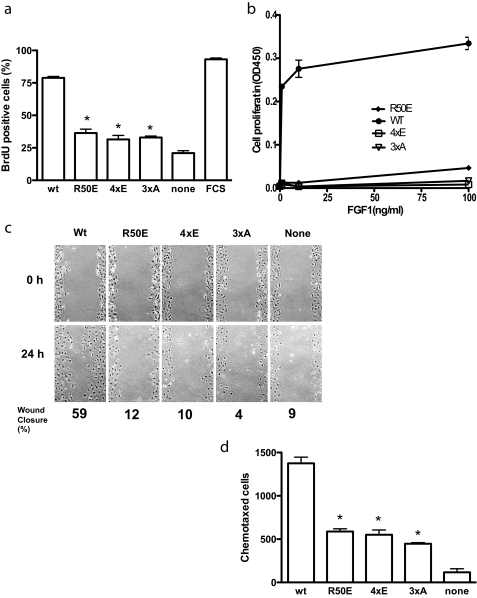
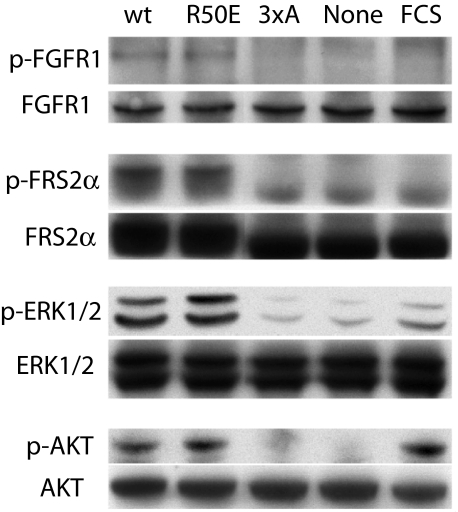
References
-
- Presta, M., Dell'Era, P., Mitola, S., Moroni, E., Ronca, R., and Rusnati, M. (2005) Cytokine Growth Factor Rev. 16 159-178 - PubMed
-
- Ullrich, A., and Schlessinger, J. (1990) Cell 61 203-212 - PubMed
-
- Powers, C. J., McLeskey, S. W., and Wellstein, A. (2000) Endocr.-Relat. Cancer 7 165-197 - PubMed
-
- Klint, P., and Claesson-Welsh, L. (1999) Front. Biosci. 4 D165-D177 - PubMed
-
- Thisse, B., and Thisse, C. (2005) Dev. Biol. 287 390-402 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
