

An efficient visualization tool for the analysis of protein mutation matrices
- PMID: 18442400
- PMCID: PMC2390542
- DOI: 10.1186/1471-2105-9-218
An efficient visualization tool for the analysis of protein mutation matrices
Abstract
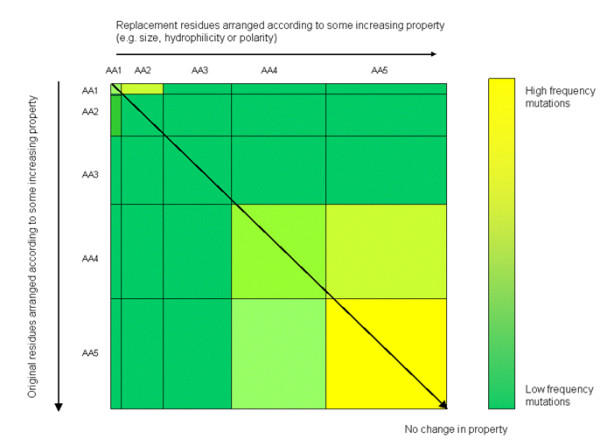
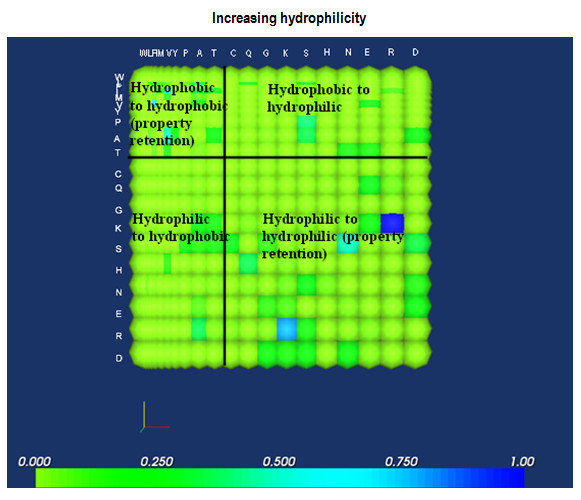
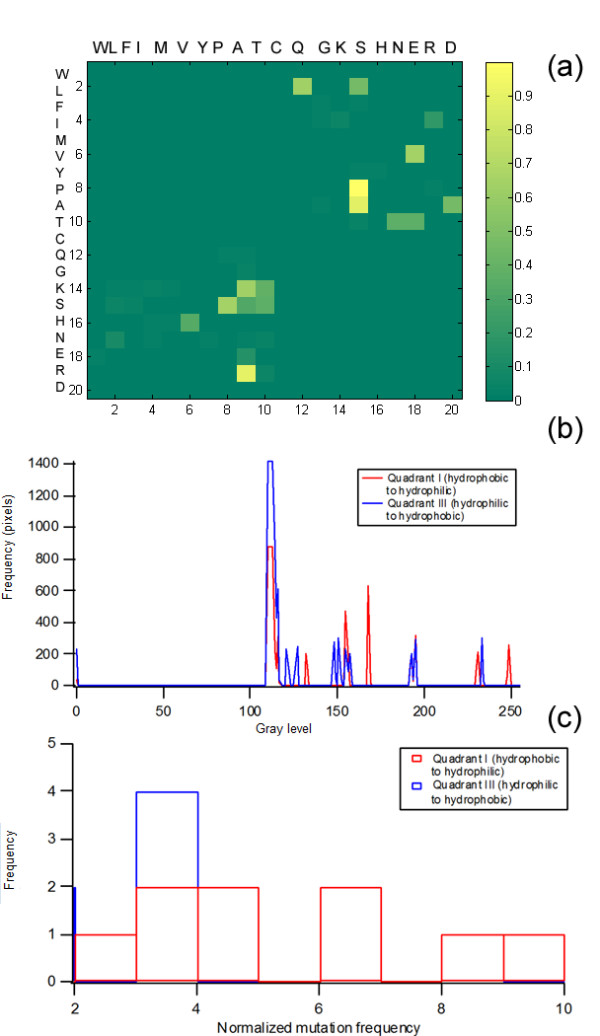
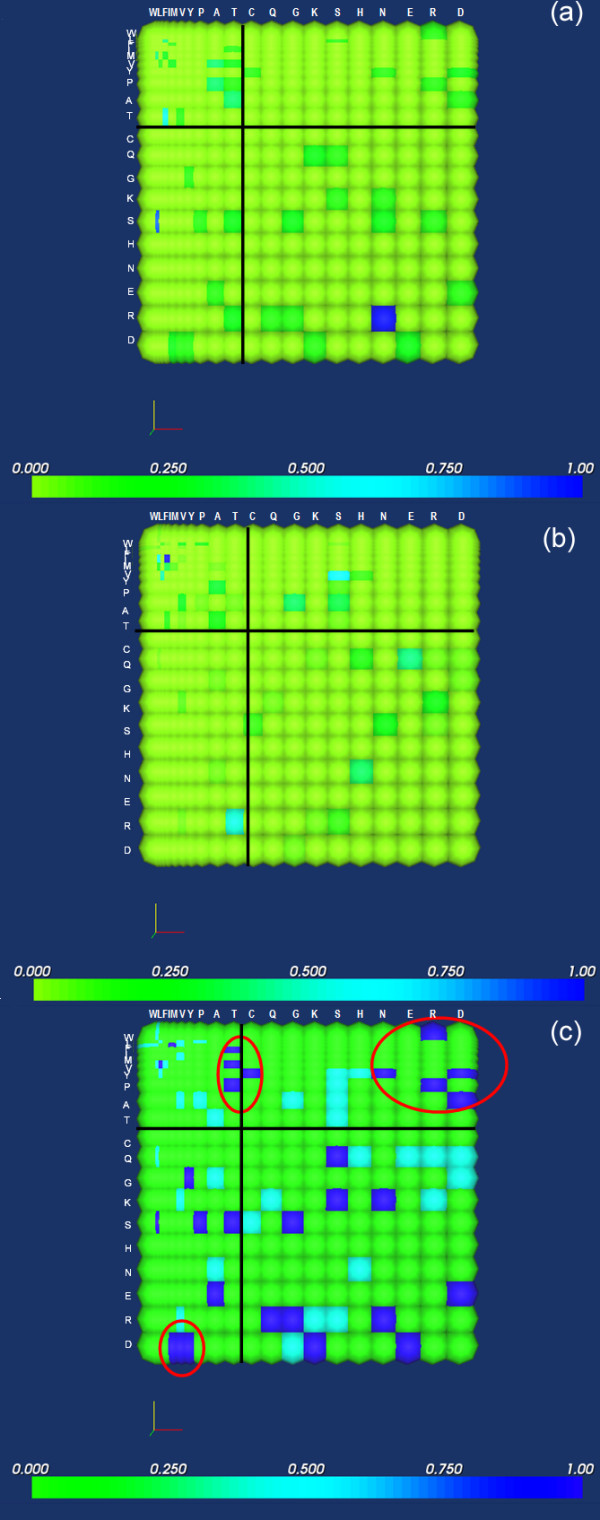
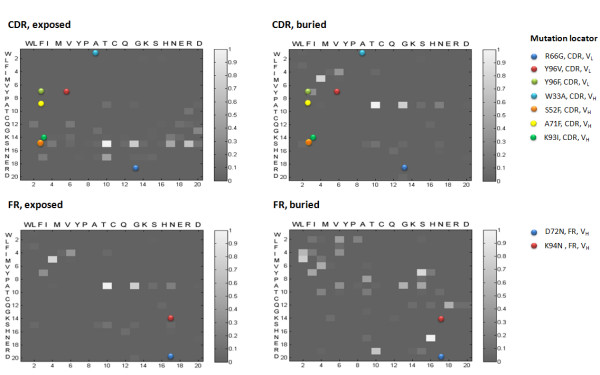
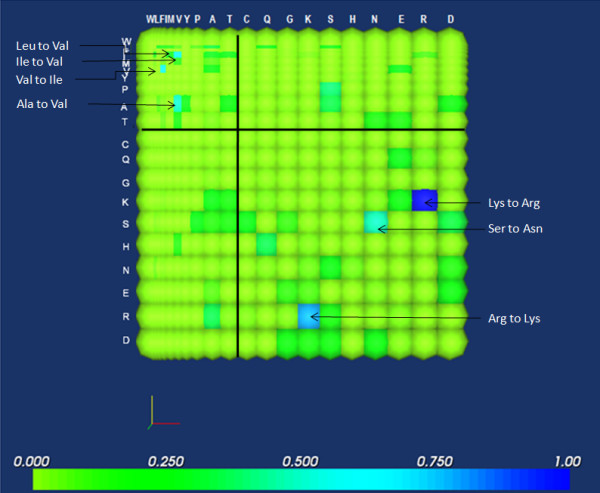
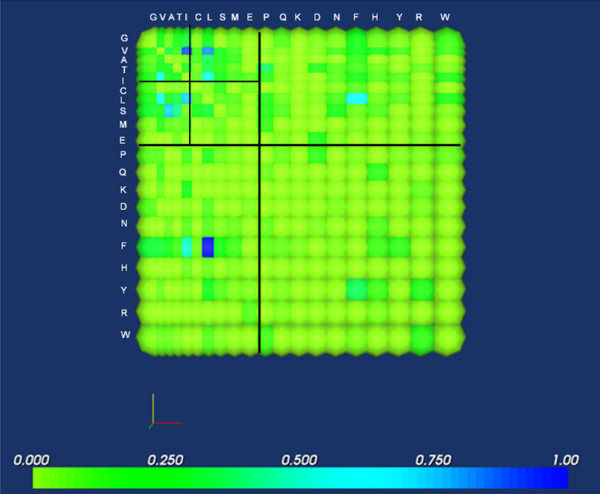
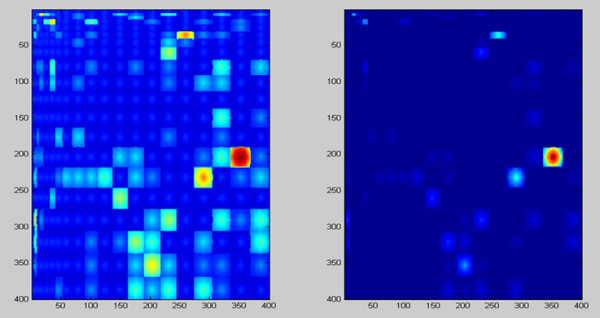
Background: It is useful to develop a tool that would effectively describe protein mutation matrices specifically geared towards the identification of mutations that produce either wanted or unwanted effects, such as an increase or decrease in affinity, or a predisposition towards misfolding. Here, we describe a tool where such mutations are efficiently identified, categorized and visualized. To categorize the mutations, amino acids in a mutation matrix are arranged according to one of three sets of physicochemical characteristics, namely hydrophilicity, size and polarizability, and charge and polarity. The magnitude and frequencies of mutations for an alignment are subsequently described using color information and scaling factors.
Results: To illustrate the capabilities of our approach, the technique is used to visualize and to compare mutation patterns in evolving sequences with diametrically opposite characteristics. Results show the emergence of distinct patterns not immediately discernible from the raw matrices.
Conclusion: Our technique enables effective categorization and visualization of mutations by using specifically-arranged mutation matrices. This tool has a number of possible applications in protein engineering, notably in simplifying the identification of mutations and/or mutation trends that are associated with specific engineered protein characteristics and behavior.
Figures


References
-
- Dayhoff MO, Schwartz RM, Orcutt BC. Atlas of protein sequence and structure. Vol. 5. National Biomedical Research Foundation, Silver Spring, MD; 1978. A model of evolutionary change in proteins.
-
- Glaser F, Pupko T, Paz I, Bell RE, Bechor D, Martz E, Ben-Tal N. ConSurf: Identification of Functional Regions in Proteins by Surface-Mapping of Phylogenetic Information. Bioinformatics. 2002;19:1–3. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
