

Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells
- PMID: 18443091
- PMCID: PMC2446742
- DOI: 10.1128/IAI.00365-08
Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells
Abstract
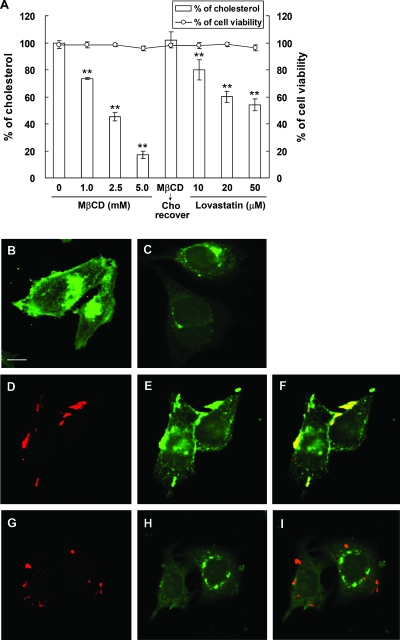
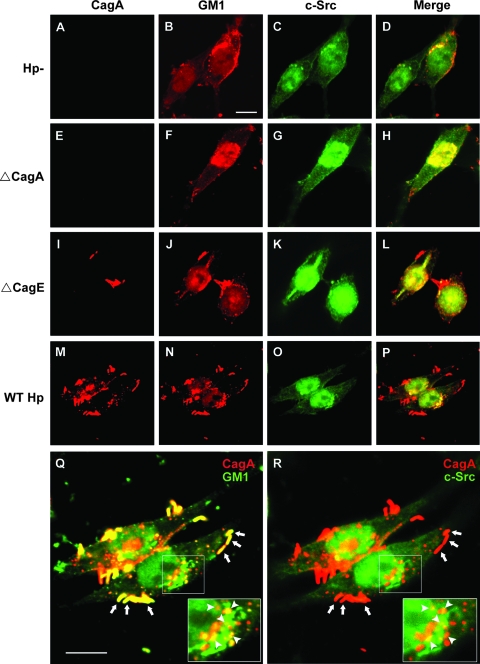
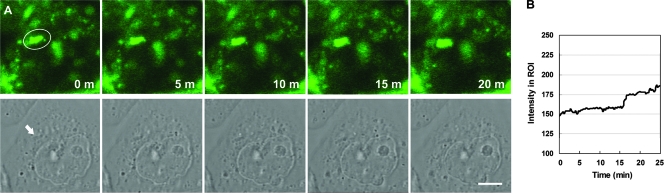
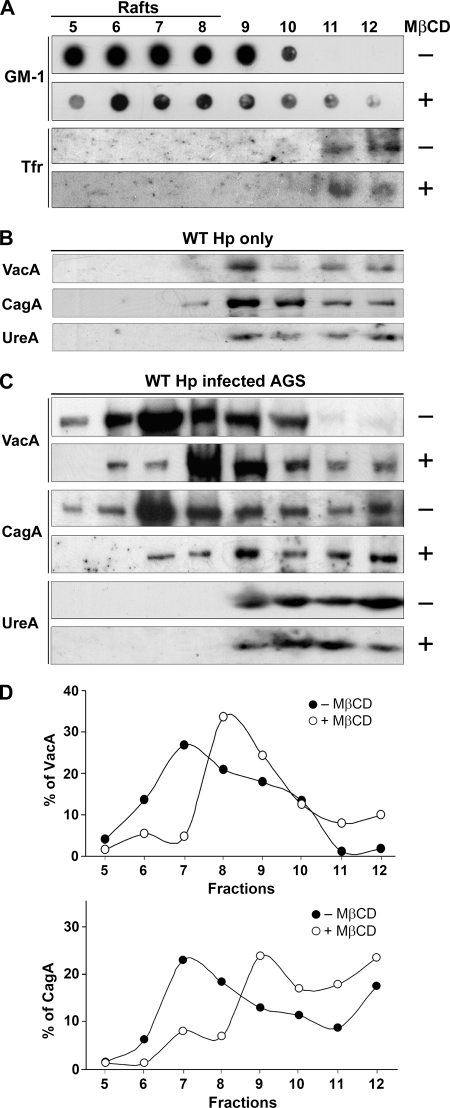
Infection with Helicobacter pylori cagA-positive strains is associated with gastritis, ulcerations, and gastric cancer. CagA is translocated into infected epithelial cells by a type IV secretion system and can be tyrosine phosphorylated, inducing signal transduction and motogenic responses in epithelial cells. Cellular cholesterol, a vital component of the membrane, contributes to membrane dynamics and functions and is important in VacA intoxication and phagocyte evasion during H. pylori infection. In this investigation, we showed that cholesterol extraction by methyl-beta-cyclodextrin reduced the level of CagA translocation and phosphorylation. Confocal microscope visualization revealed that a significant portion of translocated CagA was colocalized with the raft marker GM1 and c-Src during infection. Moreover, GM1 was rapidly recruited into sites of bacterial attachment by live-cell imaging analysis. CagA and VacA were cofractionated with detergent-resistant membranes (DRMs), suggesting that the distribution of CagA and VacA is associated with rafts in infected cells. Upon cholesterol depletion, the distribution shifted to non-DRMs. Accordingly, the CagA-induced hummingbird phenotype and interleukin-8 induction were blocked by cholesterol depletion. Raft-disrupting agents did not influence bacterial adherence but did significantly reduce internalization activity in AGS cells. Together, these results suggest that delivery of CagA into epithelial cells by the bacterial type IV secretion system is mediated in a cholesterol-dependent manner.
Figures
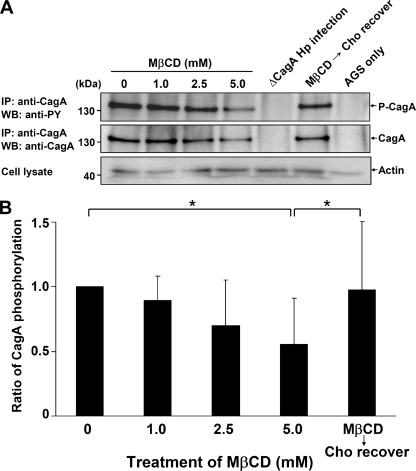

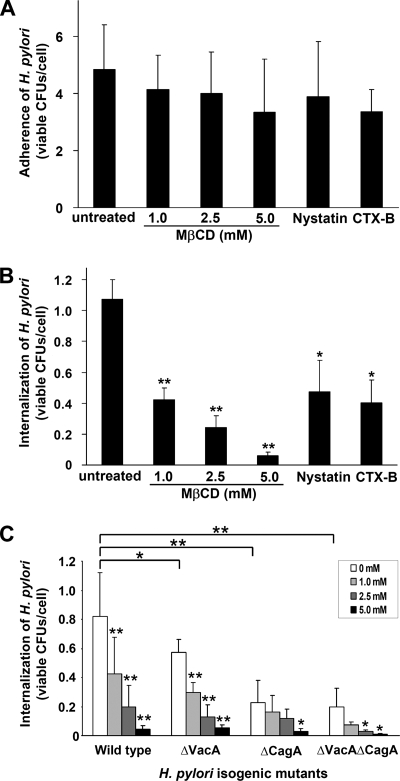
References
-
- Alm, R. A., and T. J. Trust. 1999. Analysis of the genetic diversity of Helicobacter pylori: the tale of two genomes. J. Mol. Med. 77834-846. - PubMed
-
- Amieva, M. R., N. R. Salama, L. S. Tompkins, and S. Falkow. 2002. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell. Microbiol. 4677-690. - PubMed
-
- Asahi, M., Y. Tanaka, T. Izumi, Y. Ito, H. Naiki, D. Kersulyte, K. Tsujikawa, M. Saito, K. Sada, S. Yanagi, A. Fujikawa, M. Noda, and Y. Itokawa. 2003. Helicobacter pylori CagA containing ITAM-like sequences localized to lipid rafts negatively regulates VacA-induced signaling in vivo. Helicobacter 81-14. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
