

Toward chaperone-assisted crystallography: protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold
- PMID: 18445622
- PMCID: PMC2442005
- DOI: 10.1110/ps.034892.108
Toward chaperone-assisted crystallography: protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold
Abstract
A crystallization chaperone is an auxiliary protein that binds to a target of interest, enhances and modulates crystal packing, and provides high-quality phasing information. We critically evaluated the effectiveness of a camelid single-domain antibody (V(H)H) as a crystallization chaperone. By using a yeast surface display system for V(H)H, we successfully introduced additional Met residues in the core of the V(H)H scaffold. We identified a set of SeMet-labeled V(H)H variants that collectively produced six new crystal forms as the complex with the model antigen, RNase A. The crystals exhibited monoclinic, orthorhombic, triclinic, and tetragonal symmetry and have one or two complexes in the asymmetric unit, some of which diffracted to an atomic resolution. The phasing power of the Met-enriched V(H)H chaperone allowed for auto-building the entire complex using single-anomalous dispersion technique (SAD) without the need for introducing SeMet into the target protein. We show that phases produced by combining SAD and V(H)H model-based phases are accurate enough to easily solve structures of the size reported here, eliminating the need to collect multiple wavelength multiple-anomalous dispersion (MAD) data. Together with the presence of high-throughput selection systems (e.g., phage display libraries) for V(H)H, the enhanced V(H)H domain described here will be an excellent scaffold for producing effective crystallization chaperones.
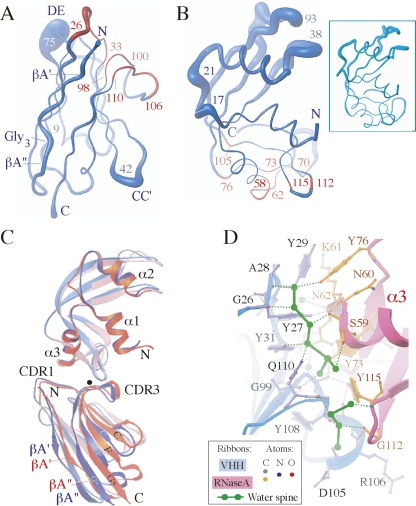
Figures
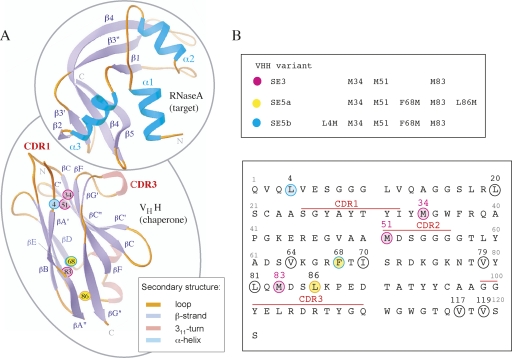
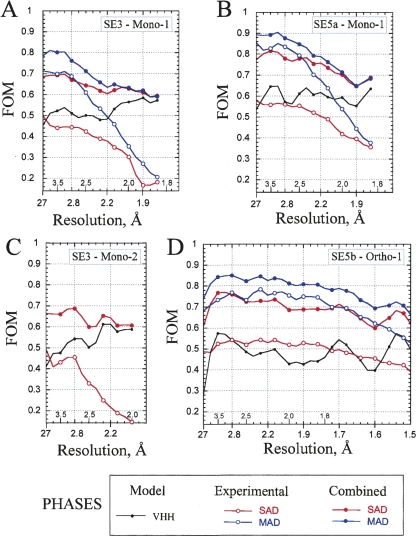
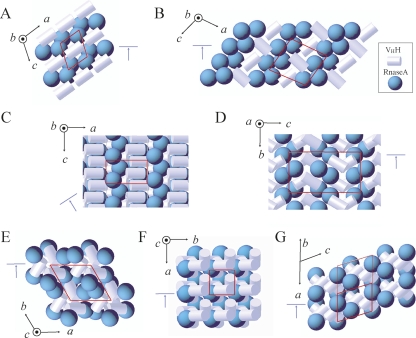
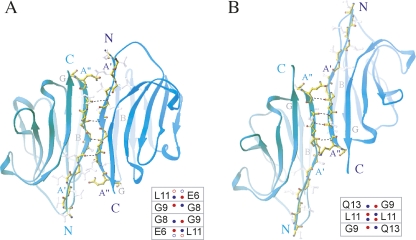
References
-
- Barthelemy, P.A., Raab, H., Appleton, B.A., Bond, C.J., Wu, P., Wiesmann, C., Sidhu, S.S. Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J. Biol. Chem. 2008;283:3639–3654. - PubMed
-
- Boder, E.T., Wittrup, K.D. Yeast surface display for directed evolution of protein expression, affinity, and stability. Methods Enzymol. 2000;328:430–444. - PubMed
-
- Bradbury, A.R.M., Marks, J.D. Antibodies from phage antibody libraries. J. Immunol. Methods. 2004;290:29–49. - PubMed
-
- Brekke, O.H., Loset, G.A. New technologies in therapeutic antibody development. Curr. Opin. Pharmacol. 2003;3:544–550. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
