

Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease
- PMID: 18449899
- PMCID: PMC2475600
- DOI: 10.1002/humu.20772
Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease
Abstract
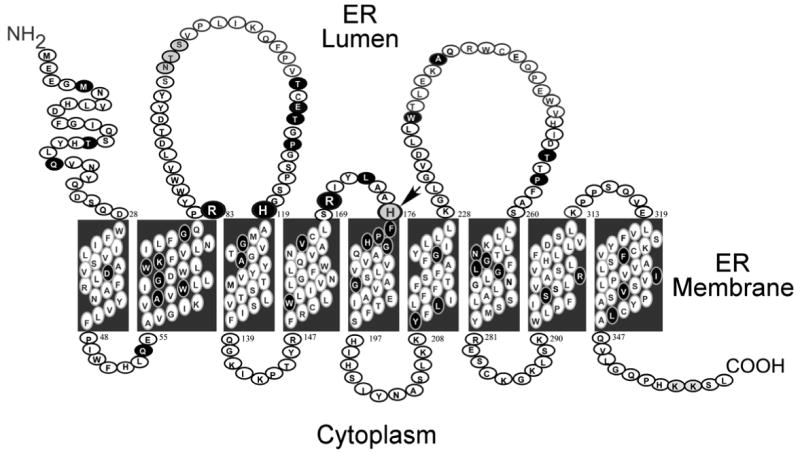
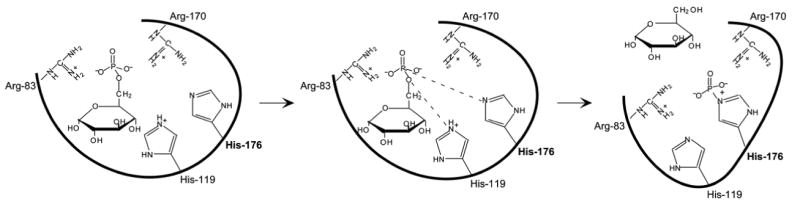
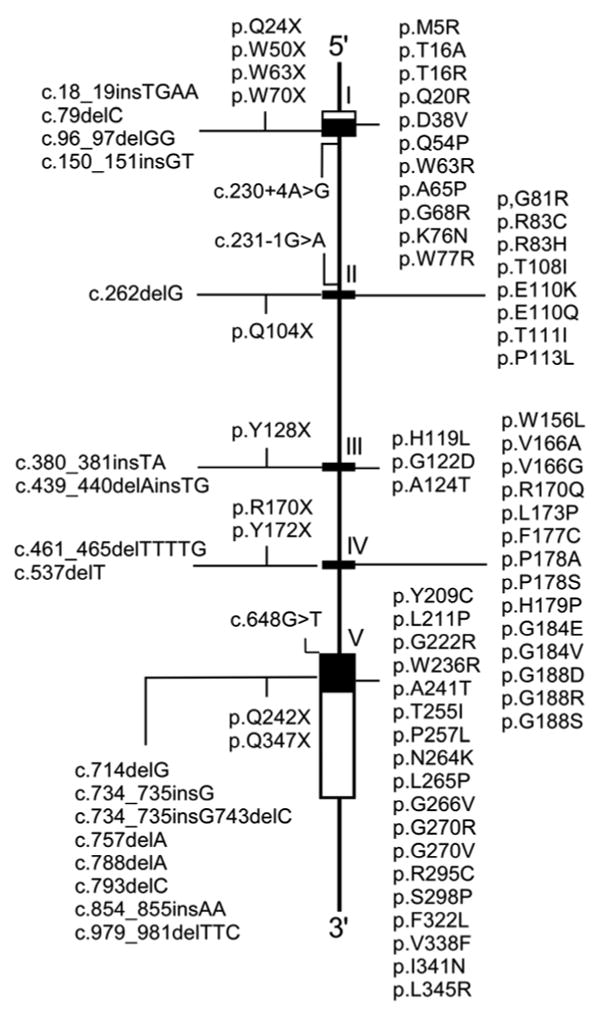
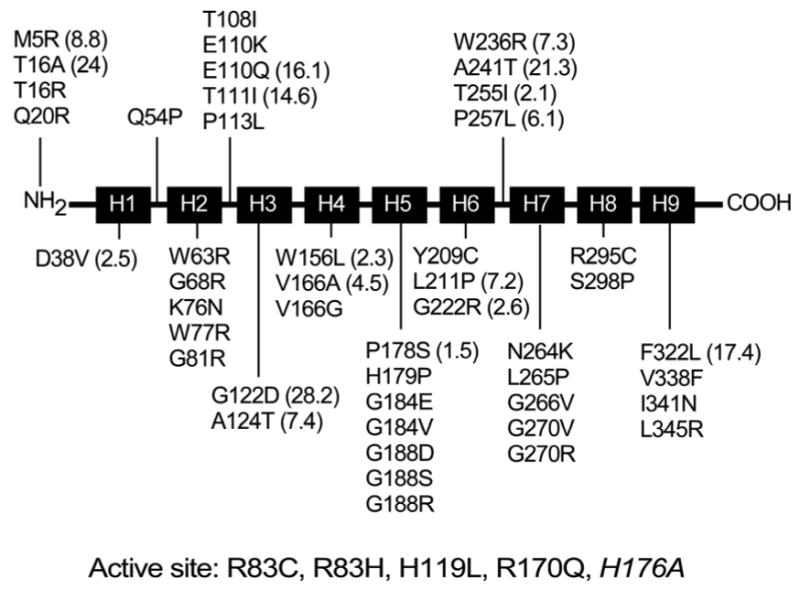
Glucose-6-phosphatase-alpha (G6PC) is a key enzyme in glucose homeostasis that catalyzes the hydrolysis of glucose-6-phosphate to glucose and phosphate in the terminal step of gluconeogenesis and glycogenolysis. Mutations in the G6PC gene, located on chromosome 17q21, result in glycogen storage disease type Ia (GSD-Ia), an autosomal recessive metabolic disorder. GSD-Ia patients manifest a disturbed glucose homeostasis, characterized by fasting hypoglycemia, hepatomegaly, nephromegaly, hyperlipidemia, hyperuricemia, lactic acidemia, and growth retardation. G6PC is a highly hydrophobic glycoprotein, anchored in the membrane of the endoplasmic reticulum with the active center facing into the lumen. To date, 54 missense, 10 nonsense, 17 insertion/deletion, and three splicing mutations in the G6PC gene have been identified in more than 550 patients. Of these, 50 missense, two nonsense, and two insertion/deletion mutations have been functionally characterized for their effects on enzymatic activity and stability. While GSD-Ia is not more prevalent in any ethnic group, mutations unique to Caucasian, Oriental, and Jewish populations have been described. Despite this, GSD-Ia patients exhibit phenotypic heterogeneity and a stringent genotype-phenotype relationship does not exist.
Figures
References
-
- Akanuma J, Nishigaki T, Fujii K, Matsubara Y, Inui K, Takahashi K, Kure S, Suzuki Y, Ohura T, Miyabayashi S, Ogawa E, Iinuma K, Okada S, Narisawa K. Glycogen storage disease type Ia: molecular diagnosis of 51 Japanese patients and characterization of slicing mutations by analysis of ectopically transcribed mRNA from lymphoblastoid cells. Am J Med Genet. 2000;91:107–112. - PubMed
-
- Angaroni CJ, Dodelson-Kremer R, Argaraña CE, Paschini-Capra AE, Giner-Ayala AN, Pezza RJ, Pan CJ, Chou JY. Glycogen Storage Disease Type Ia in Argentina. Two novel Glucose-6-phosphatase mutations affecting protein stability. Mol Genet Metab. 2004;83:276–279. - PubMed
-
- Angaroni CJ, Labrune P, Petit F, Sastre D, Capra AE, Dodelson de Kremer R, Argaraña CE. Glycogen storage disease type Ib without neutropenia generated by a novel splice-site mutation in the glucose-6-phosphate translocase gene. Mol Genet Metab. 2006;88:96–99. - PubMed
-
- Baker L, Dahlem S, Goldfarb S, Kern EF, Stanley CA, Egler J, Olshan JS, Heyman S. Hyperfiltration and renal disease in glycogen storage disease, type I. Kidney Int. 1989;35:1345–1350. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
