

Constitutive activation of the N-methyl-D-aspartate receptor via cleft-spanning disulfide bonds
- PMID: 18450751
- PMCID: PMC2490796
- DOI: 10.1074/jbc.M709190200
Constitutive activation of the N-methyl-D-aspartate receptor via cleft-spanning disulfide bonds
Abstract
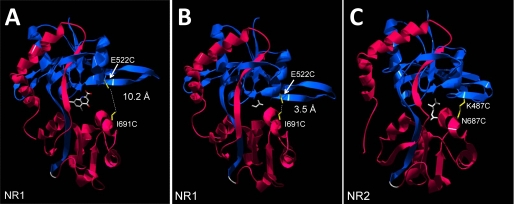
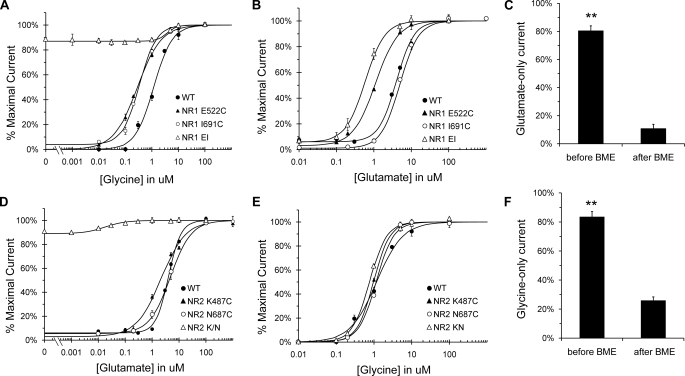
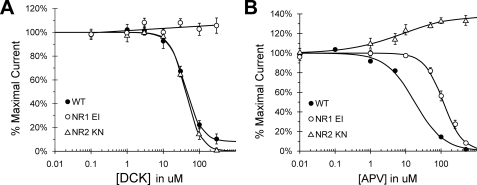
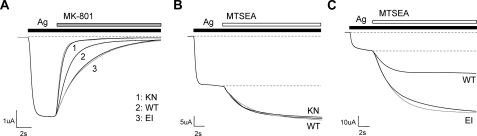
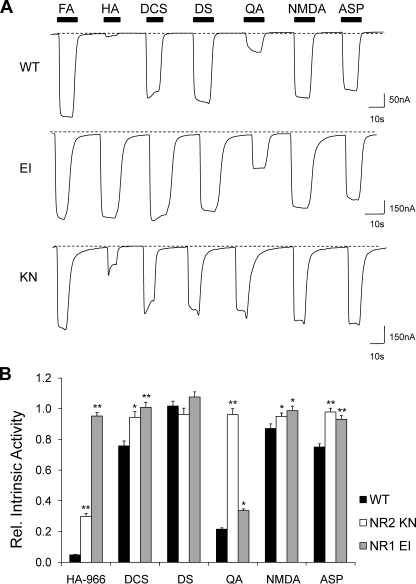
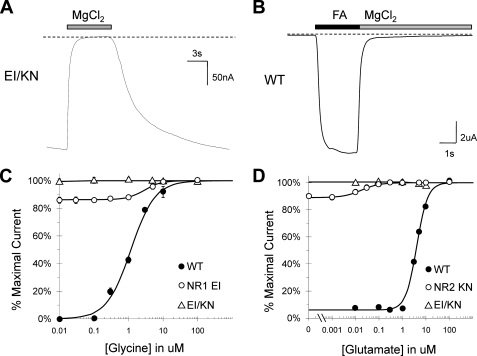
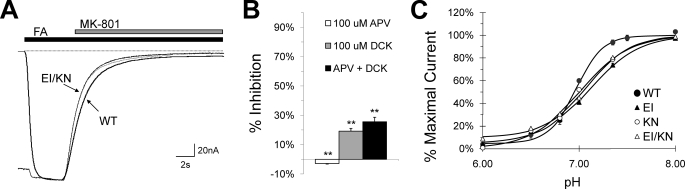
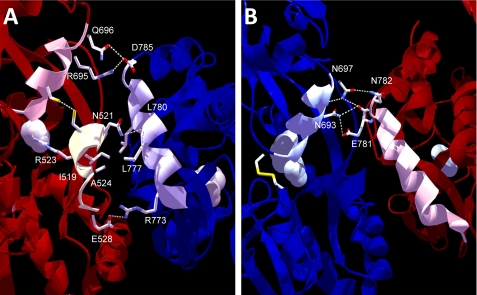
Although the N-methyl-D-aspartate (NMDA) receptor plays a critical role in the central nervous system, many questions remain regarding the relationship between its structure and functional properties. In particular, the involvement of ligand-binding domain closure in determining agonist efficacy, which has been reported in other glutamate receptor subtypes, remains unresolved. To address this question, we designed dual cysteine point mutations spanning the NR1 and NR2 ligand-binding clefts, aiming to stabilize these domains in closed cleft conformations. Two mutants, E522C/I691C in NR1 (EI) and K487C/N687C in NR2 (KN) were found to exhibit significant glycine- and glutamate-independent activation, respectively, and co-expression of the two subunits produced a constitutively active channel. However, both individual mutants could be activated above constitutive levels in a concentration-dependent manner, indicating that cleft closure does not completely prevent agonist association. Interestingly, whereas the NR2 KN disulfide was found to potentiate channel gating and M3 accessibility, NR1 EI exhibited the opposite phenotype, suggesting that the EI disulfide may trap the NR1 ligand-binding domain in a lower efficacy conformation. Furthermore, both mutants affected agonist sensitivity at the opposing subunit, suggesting that closed cleft stabilization may contribute to coupling between the subunits. These results support a correlation between cleft stability and receptor activation, providing compelling evidence for the Venus flytrap mechanism of glutamate receptor domain closure.
Figures
References
-
- Bashir, Z. I., Alford, S., Davies, S. N., Randall, A. D., and Collingridge, G. L. (1991) Nature 349 156-158 - PubMed
-
- Sheng, M., Cummings, J., Roldan, L. A., Jan, Y. N., and Jan, L. Y. (1994) Nature 368 144-147 - PubMed
-
- Kauer, J. A., Malenka, R. C., and Nicoll, R. A. (1988) Nature 334 250-252 - PubMed
-
- Hynd, M. R., Scott, H. L., and Dodd, P. R. (2004) Neurochem. Int. 45 583-595 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
