

Mapping and sequencing of structural variation from eight human genomes
- PMID: 18451855
- PMCID: PMC2424287
- DOI: 10.1038/nature06862
Mapping and sequencing of structural variation from eight human genomes
Abstract
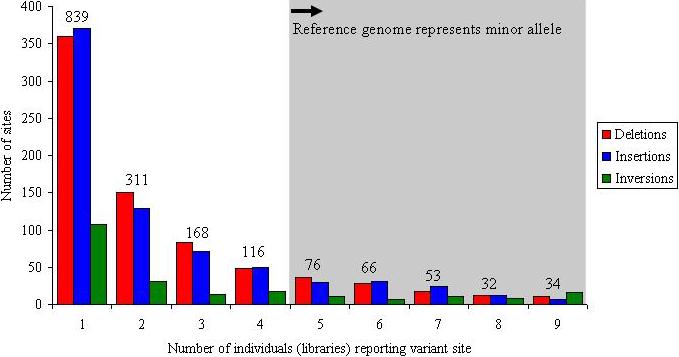
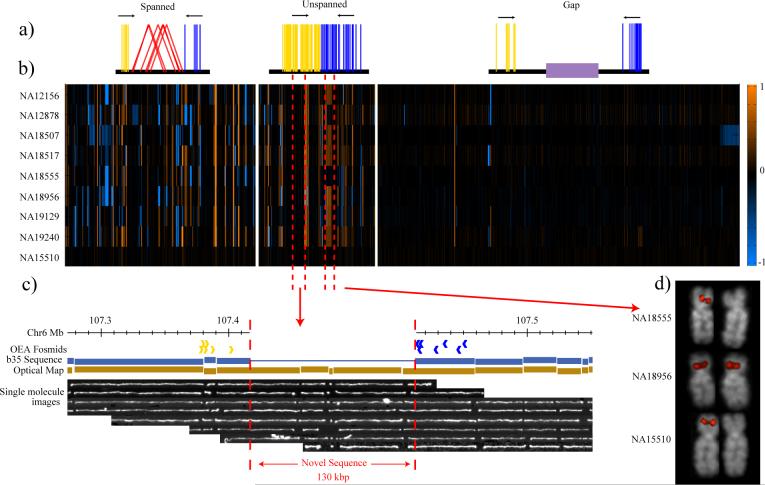
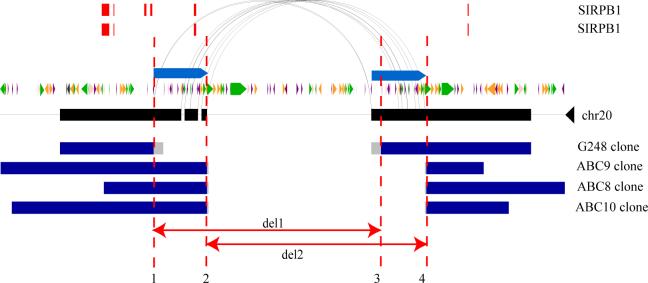
Genetic variation among individual humans occurs on many different scales, ranging from gross alterations in the human karyotype to single nucleotide changes. Here we explore variation on an intermediate scale--particularly insertions, deletions and inversions affecting from a few thousand to a few million base pairs. We employed a clone-based method to interrogate this intermediate structural variation in eight individuals of diverse geographic ancestry. Our analysis provides a comprehensive overview of the normal pattern of structural variation present in these genomes, refining the location of 1,695 structural variants. We find that 50% were seen in more than one individual and that nearly half lay outside regions of the genome previously described as structurally variant. We discover 525 new insertion sequences that are not present in the human reference genome and show that many of these are variable in copy number between individuals. Complete sequencing of 261 structural variants reveals considerable locus complexity and provides insights into the different mutational processes that have shaped the human genome. These data provide the first high-resolution sequence map of human structural variation--a standard for genotyping platforms and a prelude to future individual genome sequencing projects.
Figures


References
-
- Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nature Genet. 2004;36:949–951. - PubMed
-
- Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. - PubMed
-
- Tuzun E, et al. Fine-scale structural variation of the human genome. Nature Genet. 2005;37:727–732. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
