

Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus
- PMID: 18451997
- PMCID: PMC2350430
- DOI: 10.1172/JCI33777
Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus
Abstract
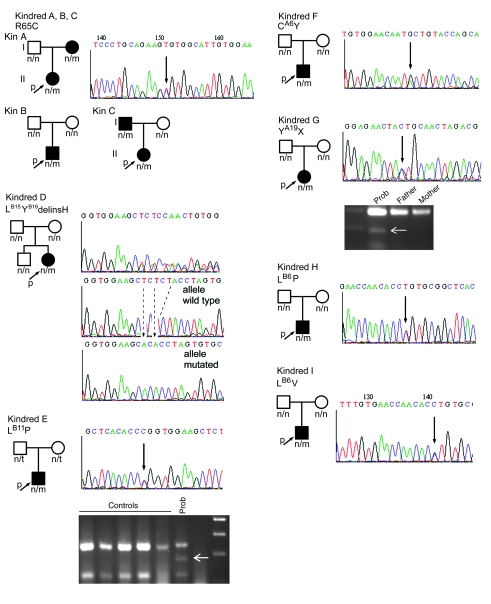
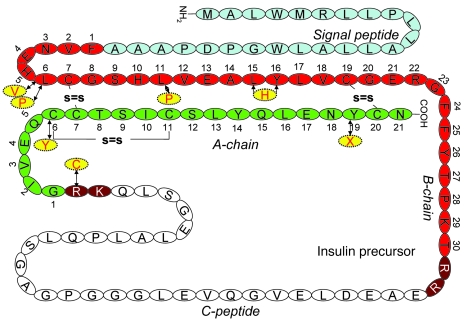
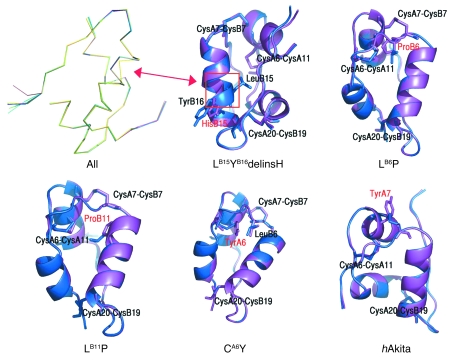
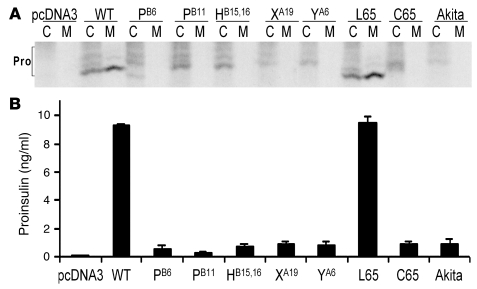
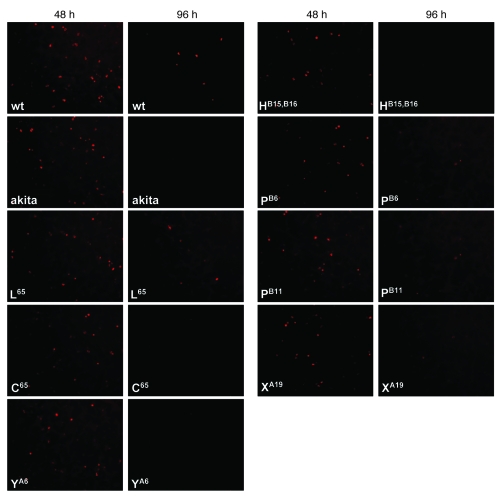
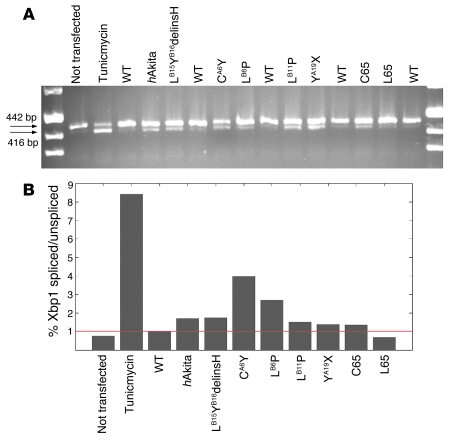
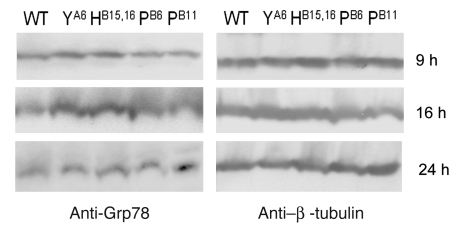
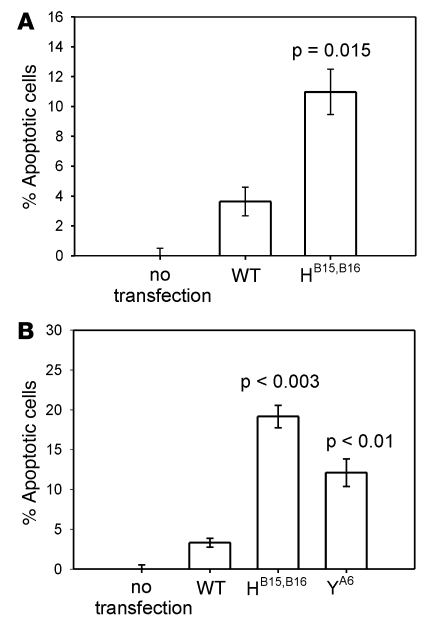
Permanent neonatal diabetes mellitus (PNDM) is a rare disorder usually presenting within 6 months of birth. Although several genes have been linked to this disorder, in almost half the cases documented in Italy, the genetic cause remains unknown. Because the Akita mouse bearing a mutation in the Ins2 gene exhibits PNDM associated with pancreatic beta cell apoptosis, we sequenced the human insulin gene in PNDM subjects with unidentified mutations. We discovered 7 heterozygous mutations in 10 unrelated probands. In 8 of these patients, insulin secretion was detectable at diabetes onset, but rapidly declined over time. When these mutant proinsulins were expressed in HEK293 cells, we observed defects in insulin protein folding and secretion. In these experiments, expression of the mutant proinsulins was also associated with increased Grp78 protein expression and XBP1 mRNA splicing, 2 markers of endoplasmic reticulum stress, and with increased apoptosis. Similarly transfected INS-1E insulinoma cells had diminished viability compared with those expressing WT proinsulin. In conclusion, we find that mutations in the insulin gene that promote proinsulin misfolding may cause PNDM.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
