

GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak
- PMID: 18451999
- PMCID: PMC2350432
- DOI: 10.1172/JCI34438
GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak
Abstract
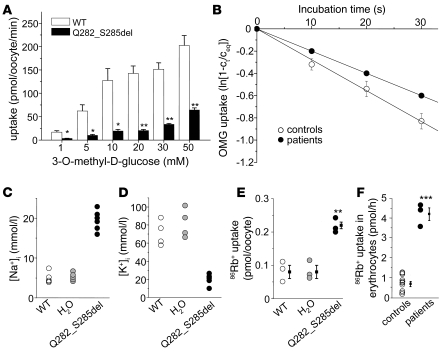
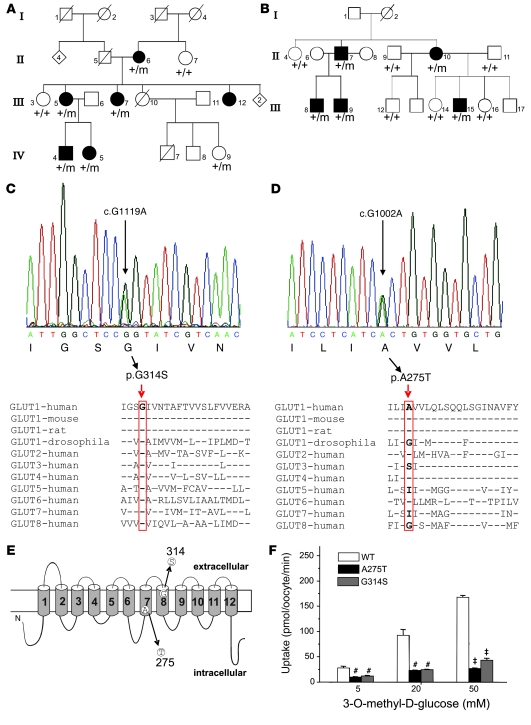
Paroxysmal dyskinesias are episodic movement disorders that can be inherited or are sporadic in nature. The pathophysiology underlying these disorders remains largely unknown but may involve disrupted ion homeostasis due to defects in cell-surface channels or nutrient transporters. In this study, we describe a family with paroxysmal exertion-induced dyskinesia (PED) over 3 generations. Their PED was accompanied by epilepsy, mild developmental delay, reduced CSF glucose levels, hemolytic anemia with echinocytosis, and altered erythrocyte ion concentrations. Using a candidate gene approach, we identified a causative deletion of 4 highly conserved amino acids (Q282_S285del) in the pore region of the glucose transporter 1 (GLUT1). Functional studies in Xenopus oocytes and human erythrocytes revealed that this mutation decreased glucose transport and caused a cation leak that alters intracellular concentrations of sodium, potassium, and calcium. We screened 4 additional families, in which PED is combined with epilepsy, developmental delay, or migraine, but not with hemolysis or echinocytosis, and identified 2 additional GLUT1 mutations (A275T, G314S) that decreased glucose transport but did not affect cation permeability. Combining these data with brain imaging studies, we propose that the dyskinesias result from an exertion-induced energy deficit that may cause episodic dysfunction of the basal ganglia, and that the hemolysis with echinocytosis may result from alterations in intracellular electrolytes caused by a cation leak through mutant GLUT1.
Figures




Similar articles
-
Stomatin-deficient cryohydrocytosis results from mutations in SLC2A1: a novel form of GLUT1 deficiency syndrome.Blood. 2011 Nov 10;118(19):5267-77. doi: 10.1182/blood-2010-12-326645. Epub 2011 Jul 26. Blood. 2011. PMID: 21791420
-
Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect.Neurology. 2011 Sep 6;77(10):959-64. doi: 10.1212/WNL.0b013e31822e0479. Epub 2011 Aug 10. Neurology. 2011. PMID: 21832227
-
Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1.Brain. 2008 Jul;131(Pt 7):1831-44. doi: 10.1093/brain/awn113. Epub 2008 Jun 24. Brain. 2008. PMID: 18577546 Free PMC article.
-
The glucose transporter type 1 (Glut1) syndromes.Epilepsy Behav. 2019 Feb;91:90-93. doi: 10.1016/j.yebeh.2018.06.010. Epub 2018 Jul 31. Epilepsy Behav. 2019. PMID: 30076047 Review.
-
The expanding phenotype of GLUT1-deficiency syndrome.Brain Dev. 2009 Aug;31(7):545-52. doi: 10.1016/j.braindev.2009.02.008. Epub 2009 Mar 21. Brain Dev. 2009. PMID: 19304421 Review.
Cited by
-
The Molecular Basis for Altered Cation Permeability in Hereditary Stomatocytic Human Red Blood Cells.Front Physiol. 2018 Apr 16;9:367. doi: 10.3389/fphys.2018.00367. eCollection 2018. Front Physiol. 2018. PMID: 29713289 Free PMC article. Review.
-
A simple blood test expedites the diagnosis of glucose transporter type 1 deficiency syndrome.Ann Neurol. 2017 Jul;82(1):133-138. doi: 10.1002/ana.24970. Ann Neurol. 2017. PMID: 28556183 Free PMC article.
-
Prevalence of inherited neurotransmitter disorders in patients with movement disorders and epilepsy: a retrospective cohort study.Orphanet J Rare Dis. 2015 Feb 8;10:12. doi: 10.1186/s13023-015-0234-9. Orphanet J Rare Dis. 2015. PMID: 25758715 Free PMC article.
-
Genetic and phenotypic heterogeneity in sporadic and familial forms of paroxysmal dyskinesia.J Neurol. 2013 Jan;260(1):93-9. doi: 10.1007/s00415-012-6592-5. Epub 2012 Jun 30. J Neurol. 2013. PMID: 22752065 Free PMC article.
-
Treatment of paroxysmal dyskinesias in children.Curr Treat Options Neurol. 2015 Jun;17(6):350. doi: 10.1007/s11940-015-0350-9. Curr Treat Options Neurol. 2015. PMID: 25864940
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
