

SCPRED: accurate prediction of protein structural class for sequences of twilight-zone similarity with predicting sequences
- PMID: 18452616
- PMCID: PMC2391167
- DOI: 10.1186/1471-2105-9-226
SCPRED: accurate prediction of protein structural class for sequences of twilight-zone similarity with predicting sequences
Abstract
Background: Protein structure prediction methods provide accurate results when a homologous protein is predicted, while poorer predictions are obtained in the absence of homologous templates. However, some protein chains that share twilight-zone pairwise identity can form similar folds and thus determining structural similarity without the sequence similarity would be desirable for the structure prediction. The folding type of a protein or its domain is defined as the structural class. Current structural class prediction methods that predict the four structural classes defined in SCOP provide up to 63% accuracy for the datasets in which sequence identity of any pair of sequences belongs to the twilight-zone. We propose SCPRED method that improves prediction accuracy for sequences that share twilight-zone pairwise similarity with sequences used for the prediction.
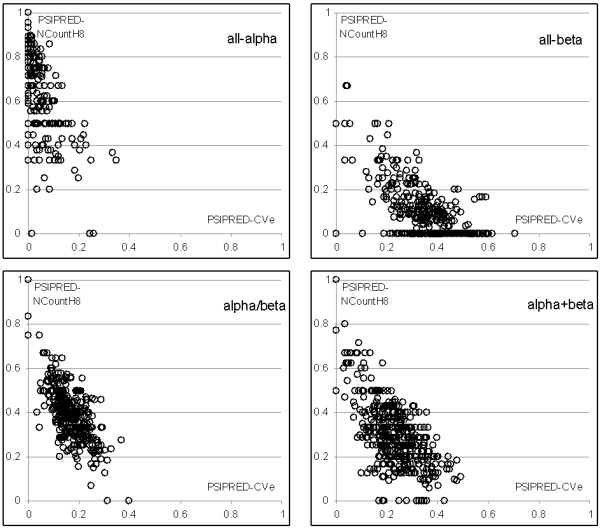
Results: SCPRED uses a support vector machine classifier that takes several custom-designed features as its input to predict the structural classes. Based on extensive design that considers over 2300 index-, composition- and physicochemical properties-based features along with features based on the predicted secondary structure and content, the classifier's input includes 8 features based on information extracted from the secondary structure predicted with PSI-PRED and one feature computed from the sequence. Tests performed with datasets of 1673 protein chains, in which any pair of sequences shares twilight-zone similarity, show that SCPRED obtains 80.3% accuracy when predicting the four SCOP-defined structural classes, which is superior when compared with over a dozen recent competing methods that are based on support vector machine, logistic regression, and ensemble of classifiers predictors.
Conclusion: The SCPRED can accurately find similar structures for sequences that share low identity with sequence used for the prediction. The high predictive accuracy achieved by SCPRED is attributed to the design of the features, which are capable of separating the structural classes in spite of their low dimensionality. We also demonstrate that the SCPRED's predictions can be successfully used as a post-processing filter to improve performance of modern fold classification methods.
Figures
Similar articles
-
Modular prediction of protein structural classes from sequences of twilight-zone identity with predicting sequences.BMC Bioinformatics. 2009 Dec 13;10:414. doi: 10.1186/1471-2105-10-414. BMC Bioinformatics. 2009. PMID: 20003388 Free PMC article.
-
Prediction of protein structural class for the twilight zone sequences.Biochem Biophys Res Commun. 2007 Jun 1;357(2):453-60. doi: 10.1016/j.bbrc.2007.03.164. Epub 2007 Apr 5. Biochem Biophys Res Commun. 2007. PMID: 17433260
-
PFRES: protein fold classification by using evolutionary information and predicted secondary structure.Bioinformatics. 2007 Nov 1;23(21):2843-50. doi: 10.1093/bioinformatics/btm475. Epub 2007 Oct 17. Bioinformatics. 2007. PMID: 17942446
-
General overview on structure prediction of twilight-zone proteins.Theor Biol Med Model. 2015 Sep 4;12:15. doi: 10.1186/s12976-015-0014-1. Theor Biol Med Model. 2015. PMID: 26338054 Free PMC article. Review.
-
Sequence and structure alignments in post-AlphaFold era.Curr Opin Struct Biol. 2023 Apr;79:102539. doi: 10.1016/j.sbi.2023.102539. Epub 2023 Feb 6. Curr Opin Struct Biol. 2023. PMID: 36753924 Review.
Cited by
-
Protein-segment universe exhibiting transitions at intermediate segment length in conformational subspaces.BMC Struct Biol. 2008 Aug 13;8:37. doi: 10.1186/1472-6807-8-37. BMC Struct Biol. 2008. PMID: 18700043 Free PMC article.
-
Prediction of Protein Structural Class Based on Gapped-Dipeptides and a Recursive Feature Selection Approach.Int J Mol Sci. 2015 Dec 24;17(1):15. doi: 10.3390/ijms17010015. Int J Mol Sci. 2015. PMID: 26712737 Free PMC article.
-
Variant Impact Predictor database (VIPdb), version 2: trends from three decades of genetic variant impact predictors.Hum Genomics. 2024 Aug 28;18(1):90. doi: 10.1186/s40246-024-00663-z. Hum Genomics. 2024. PMID: 39198917 Free PMC article.
-
Prodepth: predict residue depth by support vector regression approach from protein sequences only.PLoS One. 2009 Sep 17;4(9):e7072. doi: 10.1371/journal.pone.0007072. PLoS One. 2009. PMID: 19759917 Free PMC article.
-
VIPdb, a genetic Variant Impact Predictor Database.Hum Mutat. 2019 Sep;40(9):1202-1214. doi: 10.1002/humu.23858. Epub 2019 Aug 17. Hum Mutat. 2019. PMID: 31283070 Free PMC article.
References
-
- Chou KC. Structural bioinformatics and its impact to biomedical science. Current Medicinal Chemistry. 2004;11:2105–34. - PubMed
-
- Chou KC, Wei DQ, Du QS, Sirois S, Zhong WZ. Progress in computational approach to drug development against SARS. Current Medicinal Chemistry. 2006;13:3263–70. - PubMed
-
- Tress M, Ezkurdia I, Grana O, Lopez G, Valencia A. Assessment of predictions submitted for the CASP6 comparative modeling category. Proteins. 2005;61:27–45. - PubMed
-
- Wang G, Jin Y, Dunbrack RL., Jr Assessment of fold recognition predictions in CASP6. Proteins. 2005;61:46–66. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
