

The complete plastid genome sequence of Welwitschia mirabilis: an unusually compact plastome with accelerated divergence rates
- PMID: 18452621
- PMCID: PMC2386820
- DOI: 10.1186/1471-2148-8-130
The complete plastid genome sequence of Welwitschia mirabilis: an unusually compact plastome with accelerated divergence rates
Abstract
Background: Welwitschia mirabilis is the only extant member of the family Welwitschiaceae, one of three lineages of gnetophytes, an enigmatic group of gymnosperms variously allied with flowering plants or conifers. Limited sequence data and rapid divergence rates have precluded consensus on the evolutionary placement of gnetophytes based on molecular characters. Here we report on the first complete gnetophyte chloroplast genome sequence, from Welwitschia mirabilis, as well as analyses on divergence rates of protein-coding genes, comparisons of gene content and order, and phylogenetic implications.
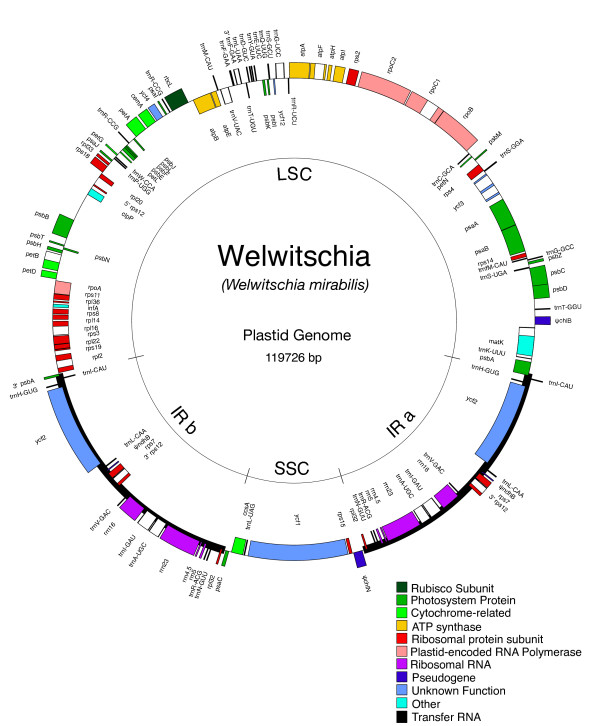
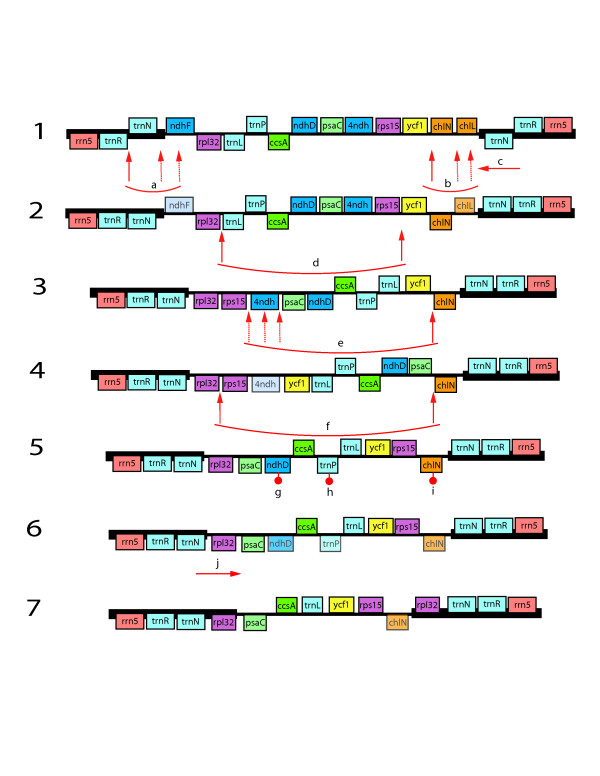
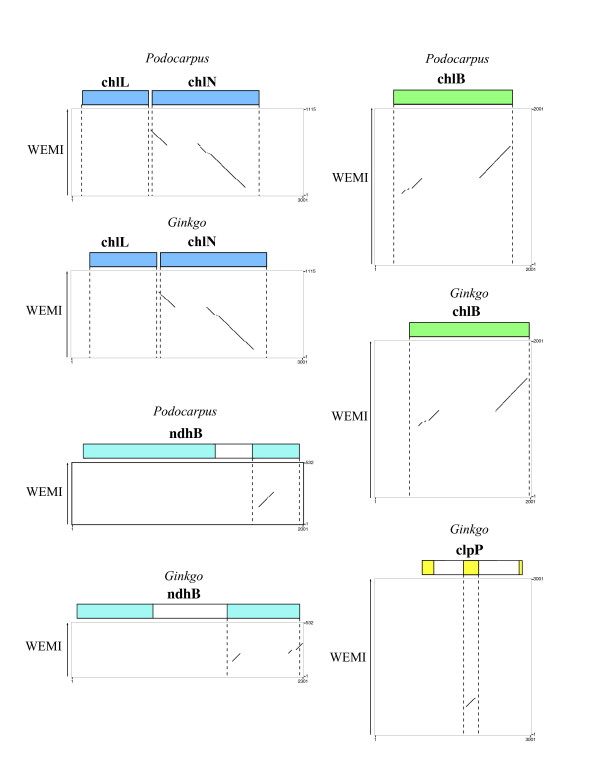
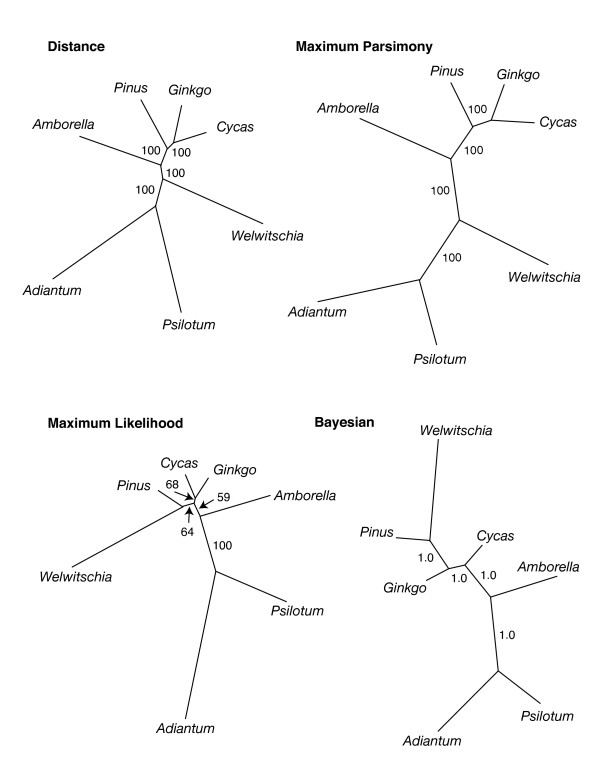
Results: The chloroplast genome of Welwitschia mirabilis [GenBank: EU342371] is comprised of 119,726 base pairs and exhibits large and small single copy regions and two copies of the large inverted repeat (IR). Only 101 unique gene species are encoded. The Welwitschia plastome is the most compact photosynthetic land plant plastome sequenced to date; 66% of the sequence codes for product. The genome also exhibits a slightly expanded IR, a minimum of 9 inversions that modify gene order, and 19 genes that are lost or present as pseudogenes. Phylogenetic analyses, including one representative of each extant seed plant lineage and based on 57 concatenated protein-coding sequences, place Welwitschia at the base of all seed plants (distance, maximum parsimony) or as the sister to Pinus (the only conifer representative) in a monophyletic gymnosperm clade (maximum likelihood, bayesian). Relative rate tests on these gene sequences show the Welwitschia sequences to be evolving at faster rates than other seed plants. For these genes individually, a comparison of average pairwise distances indicates that relative divergence in Welwitschia ranges from amounts about equal to other seed plants to amounts almost three times greater than the average for non-gnetophyte seed plants.
Conclusion: Although the basic organization of the Welwitschia plastome is typical, its compactness, gene content and high nucleotide divergence rates are atypical. The current lack of additional conifer plastome sequences precludes any discrimination between the gnetifer and gnepine hypotheses of seed plant relationships. However, both phylogenetic analyses and shared genome features identified here are consistent with either of the hypotheses that link gnetophytes with conifers, but are inconsistent with the anthophyte hypothesis.
Figures
References
-
- Gifford EM, Foster AS. Morphology and evolution of vascular plants. 3. New York: W.H. Freeman and Company; 1989.
-
- Chamberlain CJ. Gymnosperms: Structure and Function. Chicago: University of Chicago Press; 1935.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
