

Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms
- PMID: 18456842
- PMCID: PMC2390798
- DOI: 10.1261/rna.894608
Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms
Abstract
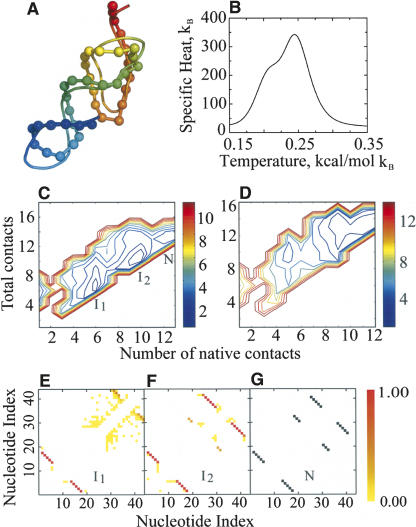
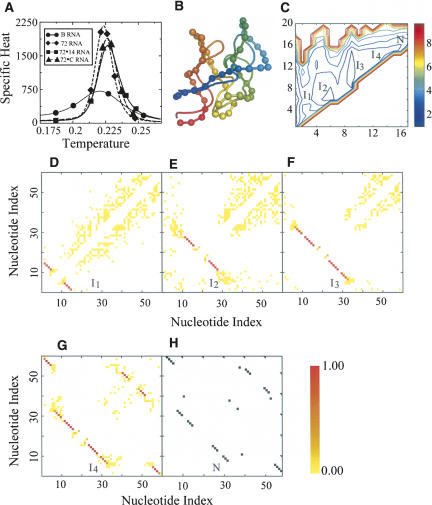
RNA molecules with novel functions have revived interest in the accurate prediction of RNA three-dimensional (3D) structure and folding dynamics. However, existing methods are inefficient in automated 3D structure prediction. Here, we report a robust computational approach for rapid folding of RNA molecules. We develop a simplified RNA model for discrete molecular dynamics (DMD) simulations, incorporating base-pairing and base-stacking interactions. We demonstrate correct folding of 150 structurally diverse RNA sequences. The majority of DMD-predicted 3D structures have <4 A deviations from experimental structures. The secondary structures corresponding to the predicted 3D structures consist of 94% native base-pair interactions. Folding thermodynamics and kinetics of tRNA(Phe), pseudoknots, and mRNA fragments in DMD simulations are in agreement with previous experimental findings. Folding of RNA molecules features transient, non-native conformations, suggesting non-hierarchical RNA folding. Our method allows rapid conformational sampling of RNA folding, with computational time increasing linearly with RNA length. We envision this approach as a promising tool for RNA structural and functional analyses.
Figures



References
-
- Andersen H.C. Molecular-dynamics simulations at constant pressure and-or temperature. J. Chem. Phys. 1980;72:2384–2393.
-
- Baumgartner A. Applications of the Monte-Carlo simulations in statistical physics. Springer; New York: 1987.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources