

ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo
- PMID: 18458112
- PMCID: PMC2373852
- DOI: 10.1084/jem.20071460
ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo
Abstract
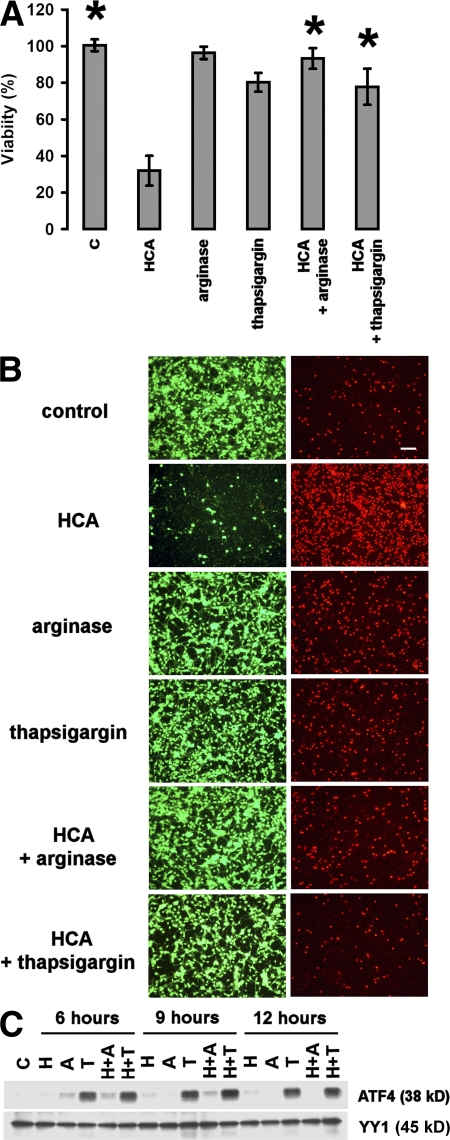
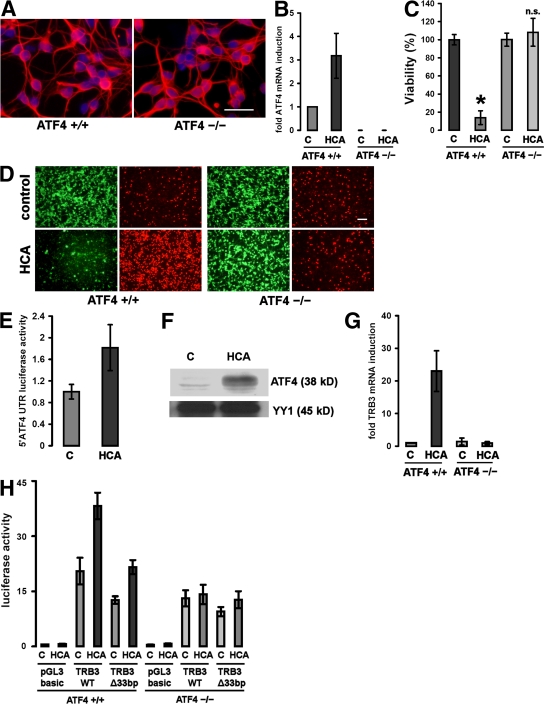
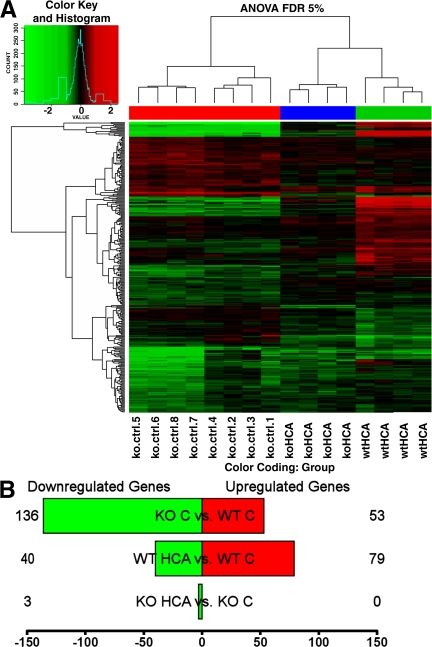
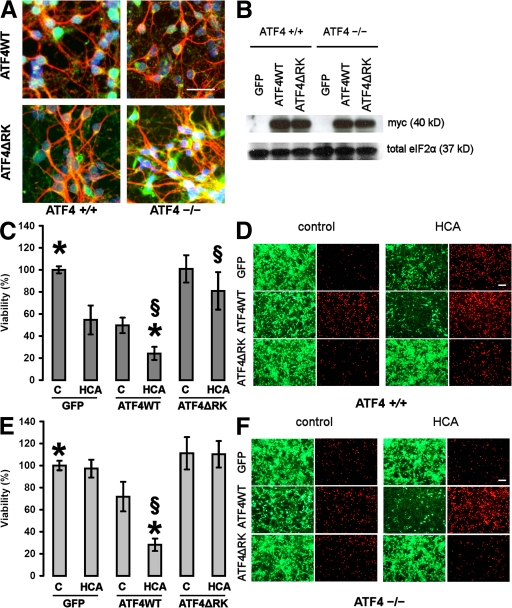
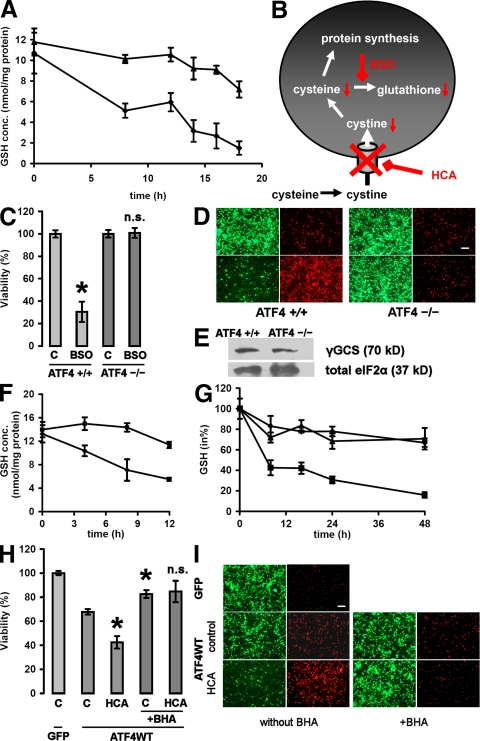
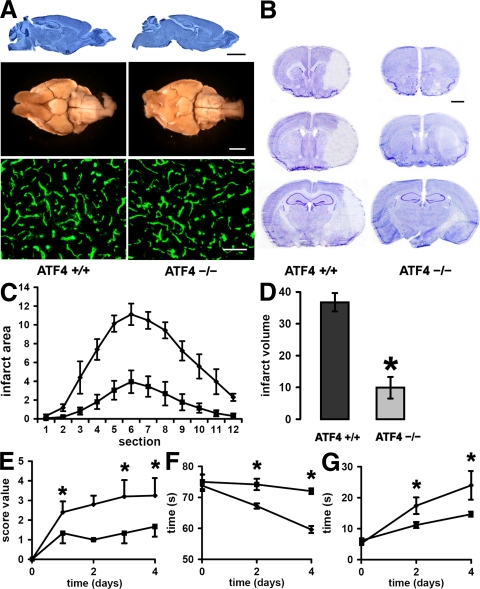
Oxidative stress is pathogenic in neurological diseases, including stroke. The identity of oxidative stress-inducible transcription factors and their role in propagating the death cascade are not well known. In an in vitro model of oxidative stress, the expression of the bZip transcription factor activating transcription factor 4 (ATF4) was induced by glutathione depletion and localized to the promoter of a putative death gene in neurons. Germline deletion of ATF4 resulted in a profound reduction in oxidative stress-induced gene expression and resistance to oxidative death. In neurons, ATF4 modulates an early, upstream event in the death pathway, as resistance to oxidative death by ATF4 deletion was associated with decreased consumption of the antioxidant glutathione. Forced expression of ATF4 was sufficient to promote cell death and loss of glutathione. In ATF4(-/-) neurons, restoration of ATF4 protein expression reinstated sensitivity to oxidative death. In addition, ATF4(-/-) mice experienced significantly smaller infarcts and improved behavioral recovery as compared with wild-type mice subjected to the same reductions in blood flow in a rodent model of ischemic stroke. Collectively, these findings establish ATF4 as a redox-regulated, prodeath transcriptional activator in the nervous system that propagates death responses to oxidative stress in vitro and to stroke in vivo.
Figures

References
-
- Droge, W. 2002. Free radicals in the physiological control of cell function. Physiol. Rev. 82:47–95. - PubMed
-
- Cullinan, S.B., and J.A. Diehl. 2006. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 38:317–332. - PubMed
-
- Beal, M.F. 2000. Oxidative metabolism. Ann. NY Acad. Sci. 924:164–169. - PubMed
-
- Banhegyi, G., A. Benedetti, M. Csala, and J. Mandl. 2007. Stress on redox. FEBS Lett. 581:3634–3640. - PubMed
-
- Veal, E.A., A.M. Day, and B.A. Morgan. 2007. Hydrogen peroxide sensing and signaling. Mol. Cell. 26:1–14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
