

Presynaptic type III neuregulin1-ErbB signaling targets {alpha}7 nicotinic acetylcholine receptors to axons
- PMID: 18458158
- PMCID: PMC2364689
- DOI: 10.1083/jcb.200710037
Presynaptic type III neuregulin1-ErbB signaling targets {alpha}7 nicotinic acetylcholine receptors to axons
Abstract
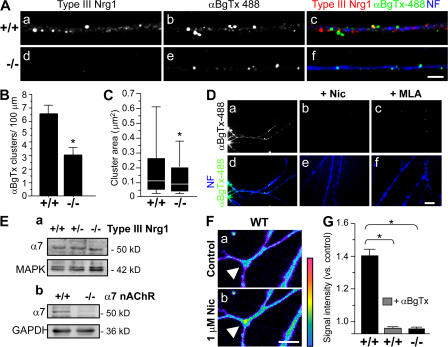
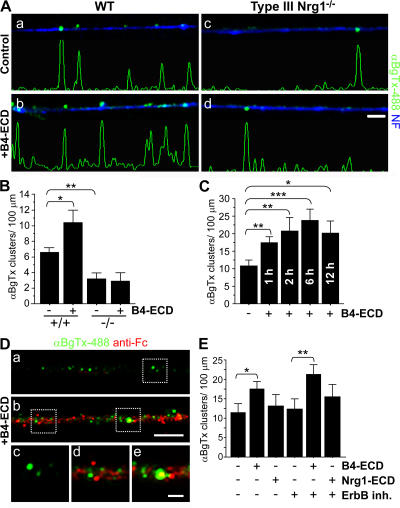
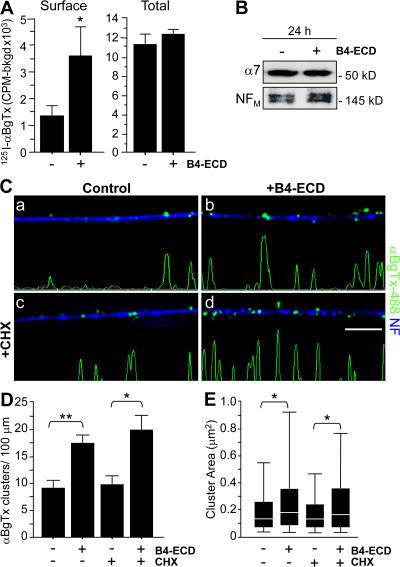
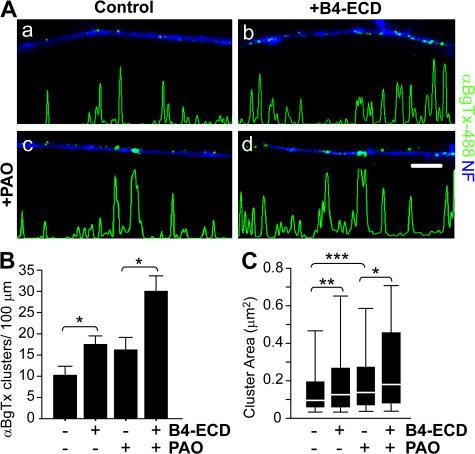
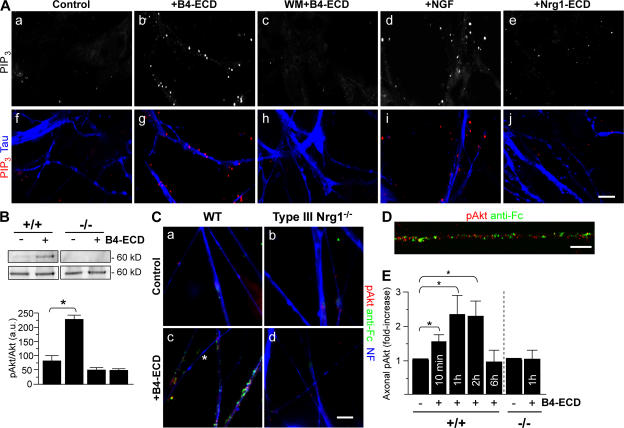
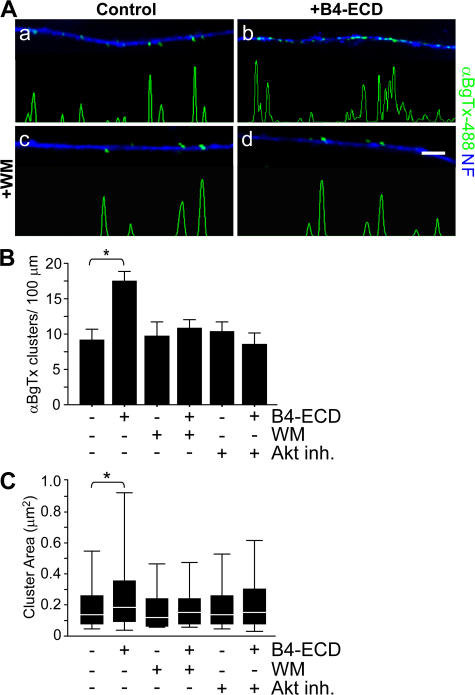
Type III Neuregulin1 (Nrg1) isoforms are membrane-tethered proteins capable of participating in bidirectional juxtacrine signaling. Neuronal nicotinic acetylcholine receptors (nAChRs), which can modulate the release of a rich array of neurotransmitters, are differentially targeted to presynaptic sites. We demonstrate that Type III Nrg1 back signaling regulates the surface expression of alpha7 nAChRs along axons of sensory neurons. Stimulation of Type III Nrg1 back signaling induces an increase in axonal surface alpha7 nAChRs, which results from a redistribution of preexisting intracellular pools of alpha7 rather than from increased protein synthesis. We also demonstrate that Type III Nrg1 back signaling activates a phosphatidylinositol 3-kinase signaling pathway and that activation of this pathway is required for the insertion of preexisting alpha7 nAChRs into the axonal plasma membrane. These findings, in conjunction with prior results establishing that Type III Nrg1 back signaling controls gene transcription, demonstrate that Type III Nrg1 back signaling can regulate both short-and long-term changes in neuronal function.
Figures
Republished in
-
Presynaptic type III neuregulin1-ErbB signaling targets alpha7 nicotinic acetylcholine receptors to axons.J Gen Physiol. 2008 Jun;131(6):i4. doi: 10.1085/JGP1316OIA4. J Gen Physiol. 2008. PMID: 18504310
References
-
- Bao, J., H. Lin, Y. Ouyang, D. Lei, A. Osman, T.W. Kim, L. Mei, P. Dai, K.K. Ohlemiller, and R.T. Ambron. 2004. Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nat. Neurosci. 7:1250–1258. - PubMed
-
- Berg, D.K., and W.G. Conroy. 2002. Nicotinic alpha 7 receptors: synaptic options and downstream signaling in neurons. J. Neurobiol. 53:512–523. - PubMed
-
- Bermingham-McDonogh, O., Y.T. Xu, M.A. Marchionni, and S.S. Scherer. 1997. Neuregulin expression in PNS neurons: isoforms and regulation by target interactions. Mol. Cell. Neurosci. 10:184–195. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
