Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome
- PMID: 18464931
- PMCID: PMC2373420
- DOI: 10.1172/JCI33578
Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome
Abstract
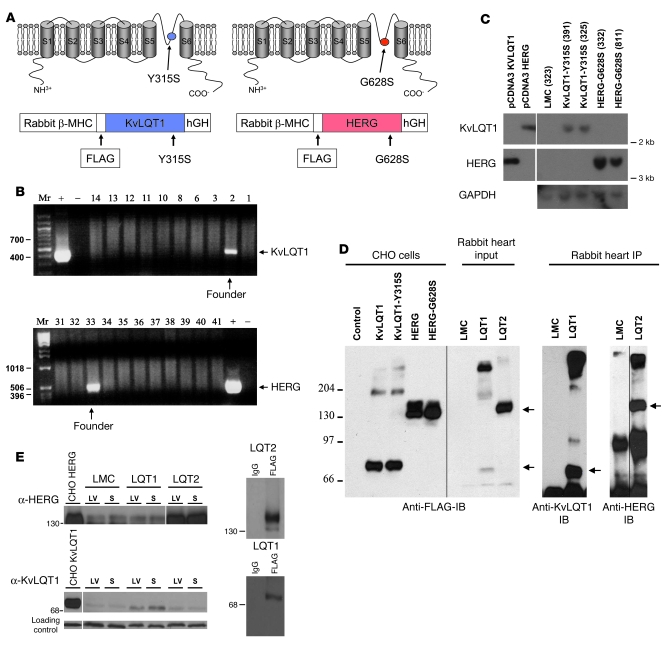
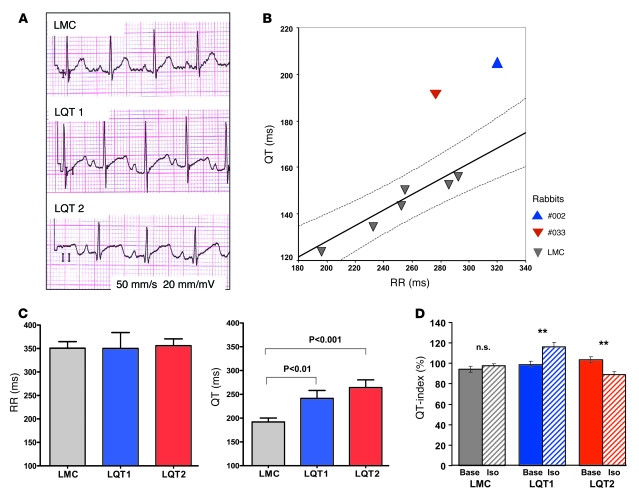
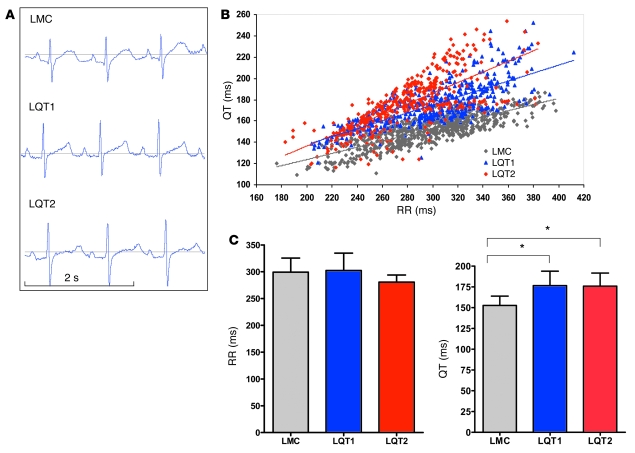
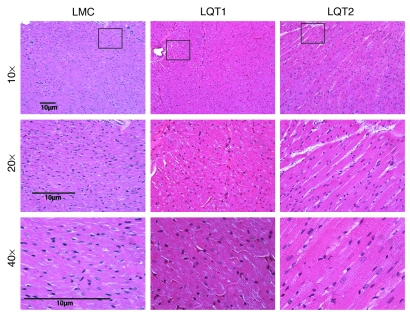
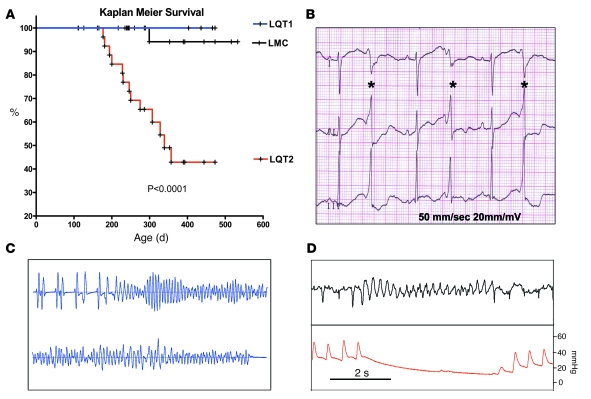
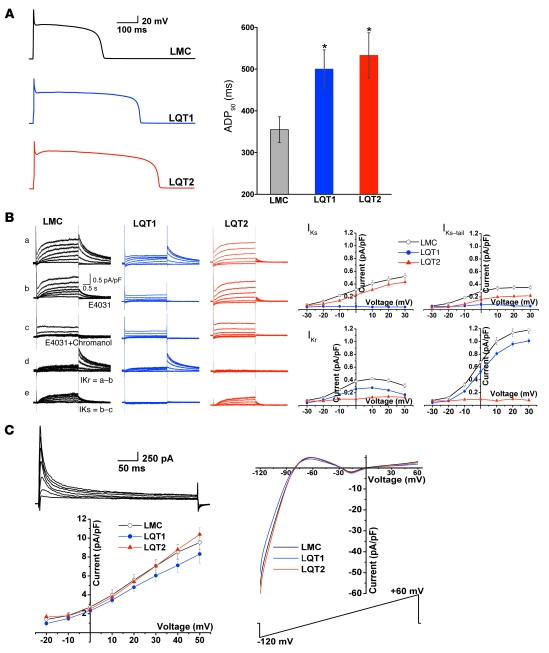
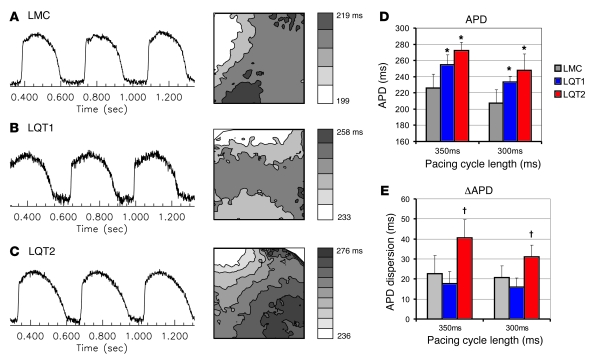
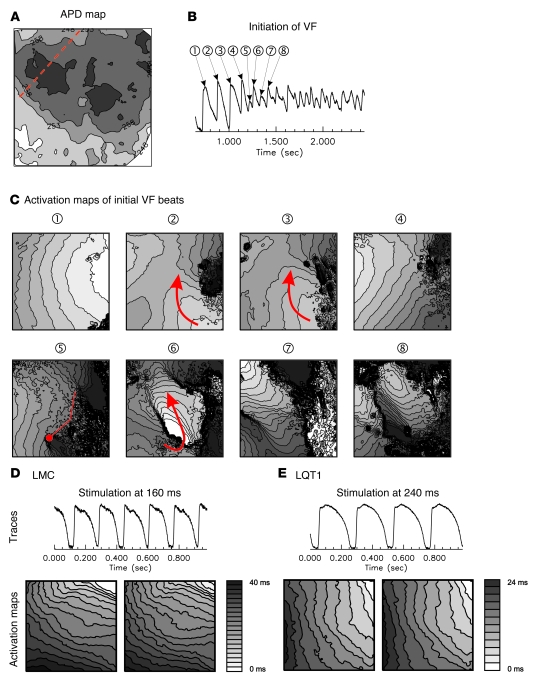
Long QT syndrome (LQTS) is a heritable disease associated with ECG QT interval prolongation, ventricular tachycardia, and sudden cardiac death in young patients. Among genotyped individuals, mutations in genes encoding repolarizing K+ channels (LQT1:KCNQ1; LQT2:KCNH2) are present in approximately 90% of affected individuals. Expression of pore mutants of the human genes KCNQ1 (KvLQT1-Y315S) and KCNH2 (HERG-G628S) in the rabbit heart produced transgenic rabbits with a long QT phenotype. Prolongations of QT intervals and action potential durations were due to the elimination of IKs and IKr currents in cardiomyocytes. LQT2 rabbits showed a high incidence of spontaneous sudden cardiac death (>50% at 1 year) due to polymorphic ventricular tachycardia. Optical mapping revealed increased spatial dispersion of repolarization underlying the arrhythmias. Both transgenes caused downregulation of the remaining complementary IKr and IKs without affecting the steady state levels of the native polypeptides. Thus, the elimination of 1 repolarizing current was associated with downregulation of the reciprocal repolarizing current rather than with the compensatory upregulation observed previously in LQTS mouse models. This suggests that mutant KvLQT1 and HERG interacted with the reciprocal wild-type alpha subunits of rabbit ERG and KvLQT1, respectively. These results have implications for understanding the nature and heterogeneity of cardiac arrhythmias and sudden cardiac death.
Figures