

Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells
- PMID: 18467441
- PMCID: PMC2488235
- DOI: 10.1210/en.2008-0117
Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells
Abstract
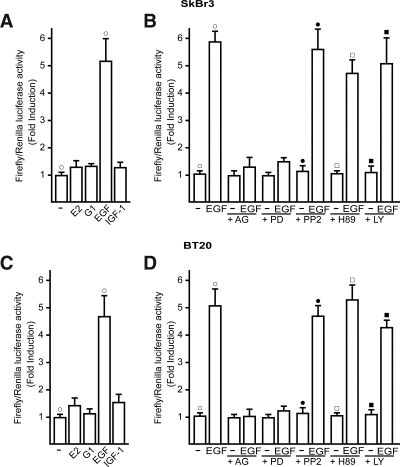
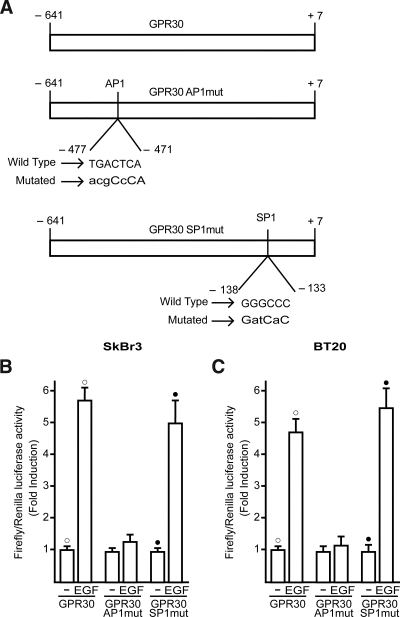
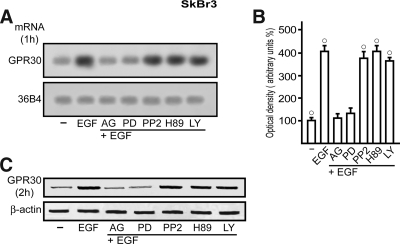
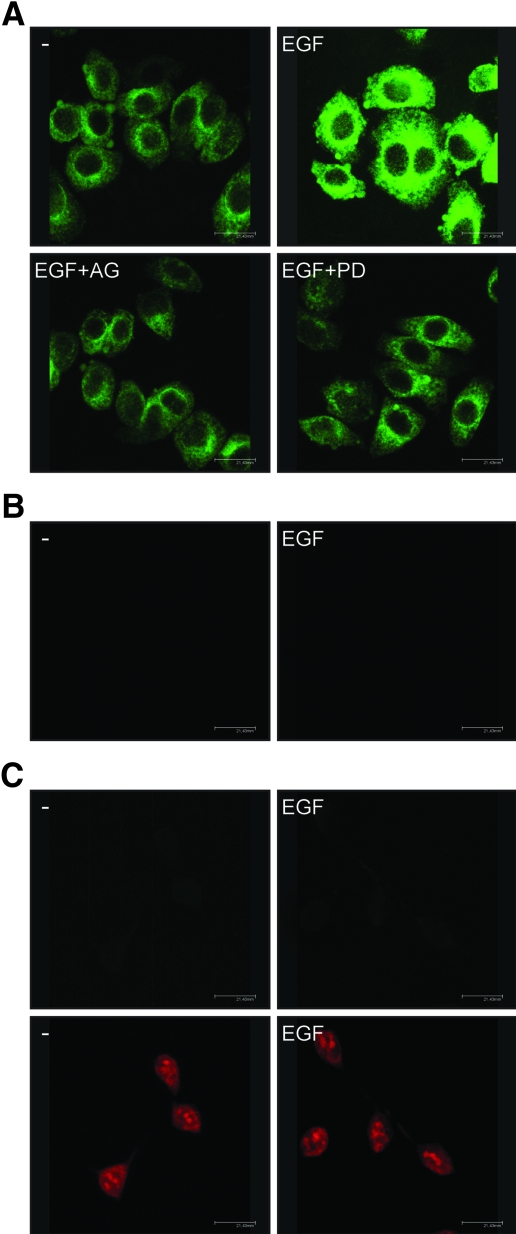
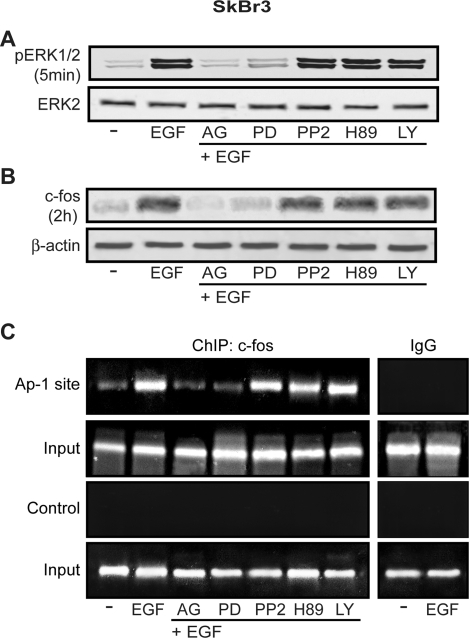
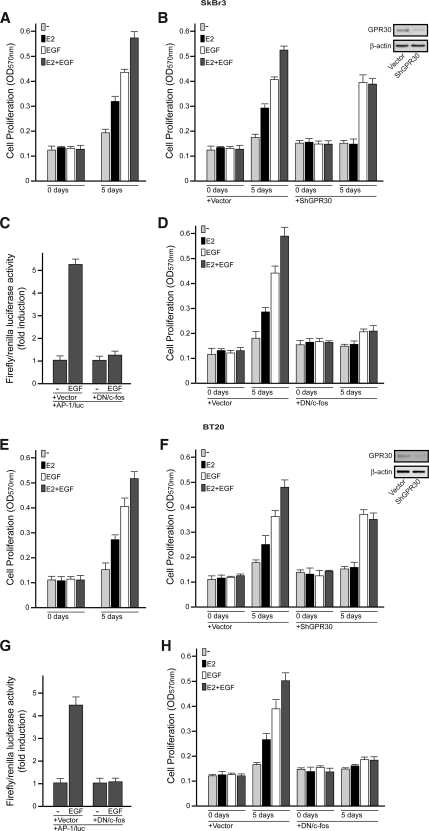
Different cellular receptors mediate the biological effects induced by estrogens. In addition to the classical nuclear estrogen receptors (ERs)-alpha and -beta, estrogen also signals through the seven-transmembrane G-protein-coupled receptor (GPR)-30. Using as a model system SkBr3 and BT20 breast cancer cells lacking the classical ER, the regulation of GPR30 expression by 17beta-estradiol, the selective GPR30 ligand G-1, IGF-I, and epidermal growth factor (EGF) was evaluated. Transient transfections with an expression plasmid encoding a short 5'-flanking sequence of the GPR30 gene revealed that an activator protein-1 site located within this region is required for the activating potential exhibited only by EGF. Accordingly, EGF up-regulated GPR30 protein levels, which accumulated predominantly in the intracellular compartment. The stimulatory role elicited by EGF on GPR30 expression was triggered through rapid ERK phosphorylation and c-fos induction, which was strongly recruited to the activator protein-1 site found in the short 5'-flanking sequence of the GPR30 gene. Of note, EGF activating the EGF receptor-MAPK transduction pathway stimulated a regulatory loop that subsequently engaged estrogen through GPR30 to boost the proliferation of SkBr3 and BT20 breast tumor cells. The up-regulation of GPR30 by ligand-activated EGF receptor-MAPK signaling provides new insight into the well-known estrogen and EGF cross talk, which, as largely reported, contributes to breast cancer progression. On the basis of our results, the action of EGF may include the up-regulation of GPR30 in facilitating a stimulatory role of estrogen, even in ER-negative breast tumor cells.
Figures

References
-
- Levin ER 2003 Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol 17:309–317 - PubMed
-
- Keen JC, Davidson NE 2003 The biology of breast carcinoma. Cancer 97:825–833 - PubMed
-
- Roskoski Jr R 2004 The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 319:1–11 - PubMed
-
- Ali S, Coombes RC 2002 Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer 2:101–112 - PubMed
-
- Razandi M, Pedram A, Park ST, Levin ER 2003 Proximal events in signaling by plasma membrane estrogen receptor. J Biol Chem 278:2701–2712 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
