

Insulin-like growth factor-II is increased in systemic sclerosis-associated pulmonary fibrosis and contributes to the fibrotic process via Jun N-terminal kinase- and phosphatidylinositol-3 kinase-dependent pathways
- PMID: 18467708
- PMCID: PMC2408418
- DOI: 10.2353/ajpath.2008.071021
Insulin-like growth factor-II is increased in systemic sclerosis-associated pulmonary fibrosis and contributes to the fibrotic process via Jun N-terminal kinase- and phosphatidylinositol-3 kinase-dependent pathways
Abstract
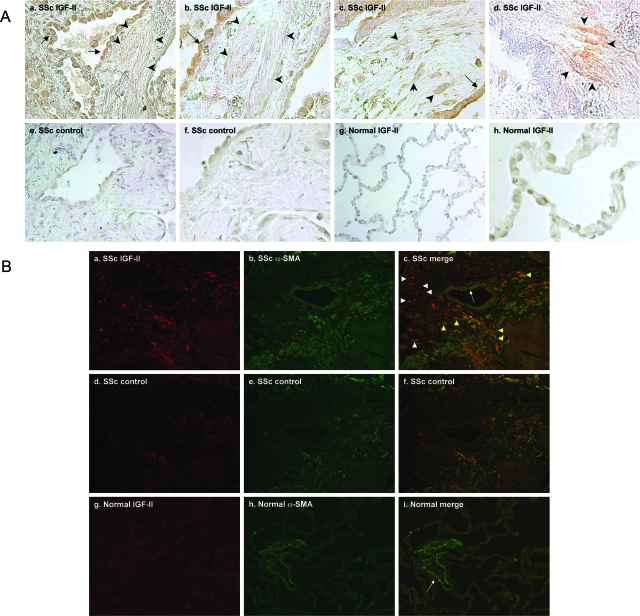
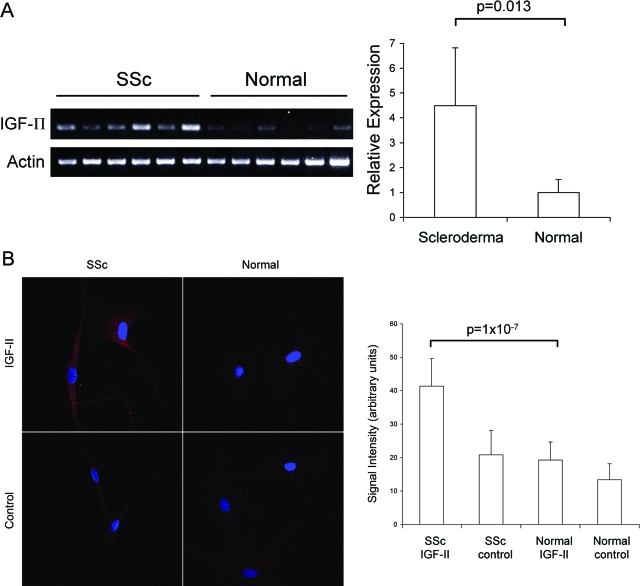
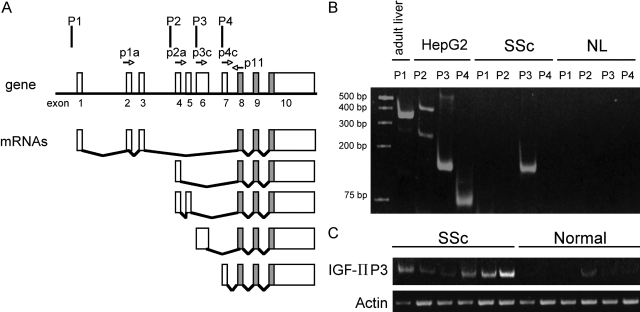
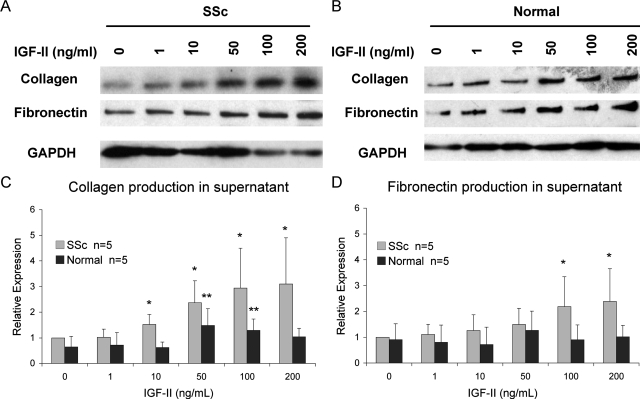
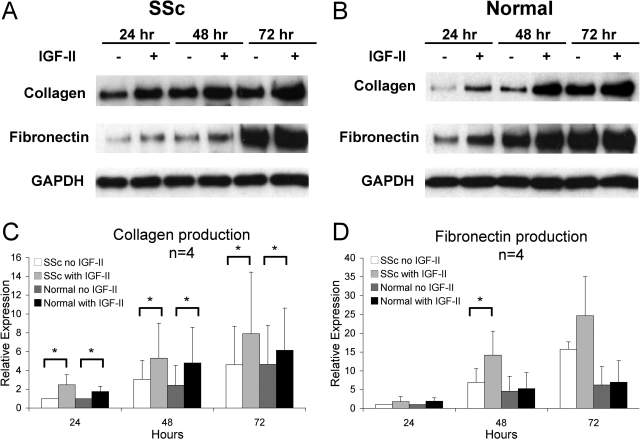
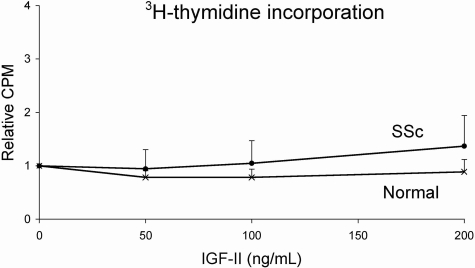
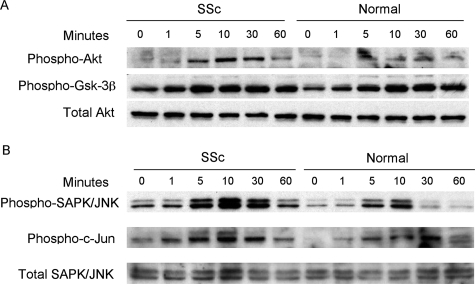
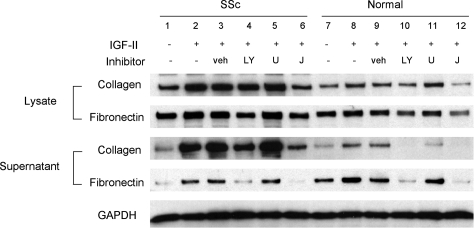
Systemic sclerosis (SSc)-related pulmonary fibrosis, for which there are few effective therapies, is the most common cause of SSc-related mortality. We examined insulin-like growth factor (IGF)-II expression in explanted lung tissues from control and SSc patients to determine its role in the pathogenesis of fibrosis. IGF-II levels in vivo were detected using immunohistochemistry. Primary lung fibroblasts were cultured from lung tissues, and IGF-II mRNA was measured using reverse transcriptase-polymerase chain reaction. Western blot analysis measured extracellular matrix (ECM) production and phosphorylated signaling molecules. Immunostaining revealed increased IGF-II expression in fibroblastic foci of SSc lungs. Furthermore, primary SSc lung fibroblasts had a fourfold increase in IGF-II mRNA and a twofold increase in IGF-II protein compared with normal lung fibroblasts. IGF-II mRNA in SSc lung fibroblasts was expressed primarily from the P3 promoter of the IGF-II gene, and IGF-II induced both a dose- and time-dependent increase in collagen type I and fibronectin production. IGF-II triggered the activation of both phosphatidylinositol-3 kinase and Jun N-terminal kinase signaling cascades, the inhibition of which diminished IGF-II-induced ECM production. Our study demonstrates increased local IGF-II expression in SSc-associated pulmonary fibrosis both in vitro and in vivo as well as IGF-II-induced ECM production through both phosphatidylinositol-3 kinase- and Jun N-terminal kinase-dependent pathways. Our results provide novel insights into the role of IGF-II in the pathogenesis of SSc-associated pulmonary fibrosis.
Figures
Similar articles
-
Localized expression of tenascin in systemic sclerosis-associated pulmonary fibrosis and its regulation by insulin-like growth factor binding protein 3.Arthritis Rheum. 2012 Jan;64(1):272-80. doi: 10.1002/art.30647. Arthritis Rheum. 2012. PMID: 21898349 Free PMC article.
-
Insulin-like growth factor (IGF)-II- mediated fibrosis in pathogenic lung conditions.PLoS One. 2019 Nov 25;14(11):e0225422. doi: 10.1371/journal.pone.0225422. eCollection 2019. PLoS One. 2019. PMID: 31765403 Free PMC article.
-
The Role of SOX9 in IGF-II-Mediated Pulmonary Fibrosis.Int J Mol Sci. 2023 Jul 8;24(14):11234. doi: 10.3390/ijms241411234. Int J Mol Sci. 2023. PMID: 37510994 Free PMC article.
-
Autocrine TGF-beta signaling in the pathogenesis of systemic sclerosis.J Dermatol Sci. 2008 Feb;49(2):103-13. doi: 10.1016/j.jdermsci.2007.05.014. Epub 2007 Jul 12. J Dermatol Sci. 2008. PMID: 17628443 Review.
-
Scleroderma, fibroblasts, signaling, and excessive extracellular matrix.Curr Rheumatol Rep. 2005 Apr;7(2):156-62. doi: 10.1007/s11926-005-0069-9. Curr Rheumatol Rep. 2005. PMID: 15760596 Review.
Cited by
-
Prominence of IL6, IGF, TLR, and Bioenergetics Pathway Perturbation in Lung Tissues of Scleroderma Patients With Pulmonary Fibrosis.Front Immunol. 2020 Mar 10;11:383. doi: 10.3389/fimmu.2020.00383. eCollection 2020. Front Immunol. 2020. PMID: 32210969 Free PMC article.
-
A review of recent studies on the pathogenesis of Systemic Sclerosis: focus on fibrosis pathways.Front Immunol. 2025 Apr 16;16:1551911. doi: 10.3389/fimmu.2025.1551911. eCollection 2025. Front Immunol. 2025. PMID: 40308583 Free PMC article. Review.
-
First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry.Int J Mol Sci. 2023 Feb 11;24(4):3645. doi: 10.3390/ijms24043645. Int J Mol Sci. 2023. PMID: 36835058 Free PMC article.
-
MRI Assessment of Treatment Response in HIV-associated NAFLD: A Randomized Trial of a Stearoyl-Coenzyme-A-Desaturase-1 Inhibitor (ARRIVE Trial).Hepatology. 2019 Nov;70(5):1531-1545. doi: 10.1002/hep.30674. Epub 2019 Jun 18. Hepatology. 2019. PMID: 31013363 Free PMC article. Clinical Trial.
-
The fibrotic phenotype induced by IGFBP-5 is regulated by MAPK activation and egr-1-dependent and -independent mechanisms.Am J Pathol. 2009 Aug;175(2):605-15. doi: 10.2353/ajpath.2009.080991. Epub 2009 Jul 23. Am J Pathol. 2009. PMID: 19628764 Free PMC article.
References
-
- Harrison NK, Cambrey AD, Myers AR, Southcott AM, Black CM, du Bois RM, Laurent GJ, McAnulty RJ. Insulin-like growth factor-I is partially responsible for fibroblast proliferation induced by bronchoalveolar lavage fluid from patients with systemic sclerosis. Clin Sci (Lond) 1994;86:141–148. - PubMed
-
- Homma S, Nagaoka I, Abe H, Takahashi K, Seyama K, Nukiwa T, Kira S. Localization of platelet-derived growth factor and insulin-like growth factor I in the fibrotic lung. Am J Respir Crit Care Med. 1995;152:2084–2089. - PubMed
-
- Vanhee D, Gosset P, Wallaert B, Voisin C, Tonnel AB. Mechanisms of fibrosis in coal workers’ pneumoconiosis. Increased production of platelet-derived growth factor, insulin-like growth factor type I, and transforming growth factor beta and relationship to disease severity. Am J Respir Crit Care Med. 1994;150:1049–1055. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
