

Phosphorylation of Bad at Thr-201 by JNK1 promotes glycolysis through activation of phosphofructokinase-1
- PMID: 18469002
- PMCID: PMC2475697
- DOI: 10.1074/jbc.M800024200
Phosphorylation of Bad at Thr-201 by JNK1 promotes glycolysis through activation of phosphofructokinase-1
Abstract
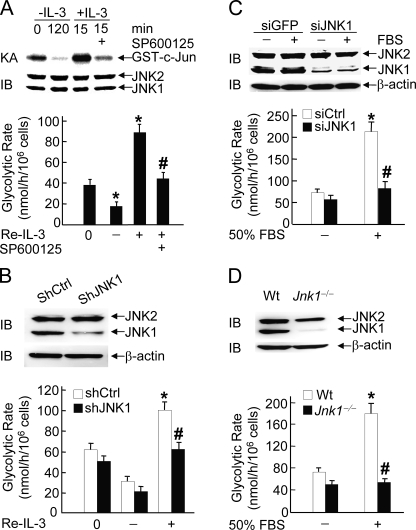
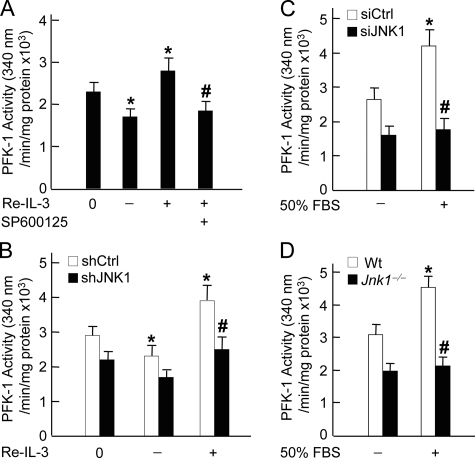
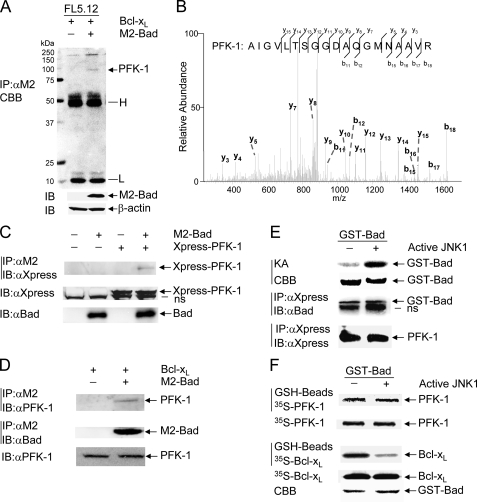
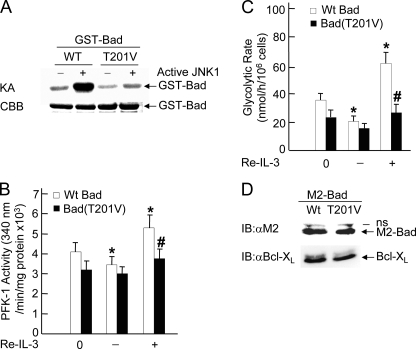
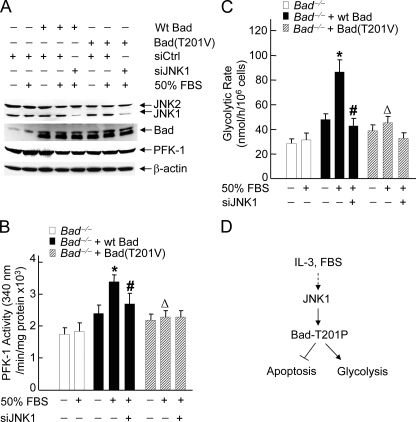
The mitogen-activated protein kinase JNK1 suppresses interleukin-3 withdrawal-induced cell death through phosphorylation of the BH3-only pro-apoptotic Bcl-2 family protein Bad at Thr-201. It is unknown whether JNK1 regulates glycolysis, an important metabolic process that is involved in cell survival, and if so, whether the regulation depends on Thr-201 phosphorylation of Bad. Here we report that phosphorylation of Bad by JNK1 is required for glycolysis through activation of phosphofructokinase-1 (PFK-1), one of the key enzymes that catalyze glycolysis. Genetic disruption of Jnk1 alleles or silencing of Jnk1 by small interfering RNA abrogates glycolysis induced by growth/survival factors such as serum or interleukin-3. Proteomic analysis identifies PFK-1 as a novel Bad-associated protein. Although the interaction between PFK-1 and Bad is independent of JNK1, Thr-201 phosphorylation of Bad by JNK1 is required for PFK-1 activation. Thus, our results provide a novel molecular mechanism by which JNK1 promotes glycolysis for cell survival.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
