

Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links
- PMID: 18469862
- PMCID: PMC2805112
- DOI: 10.1038/onc.2008.139
Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links
Abstract
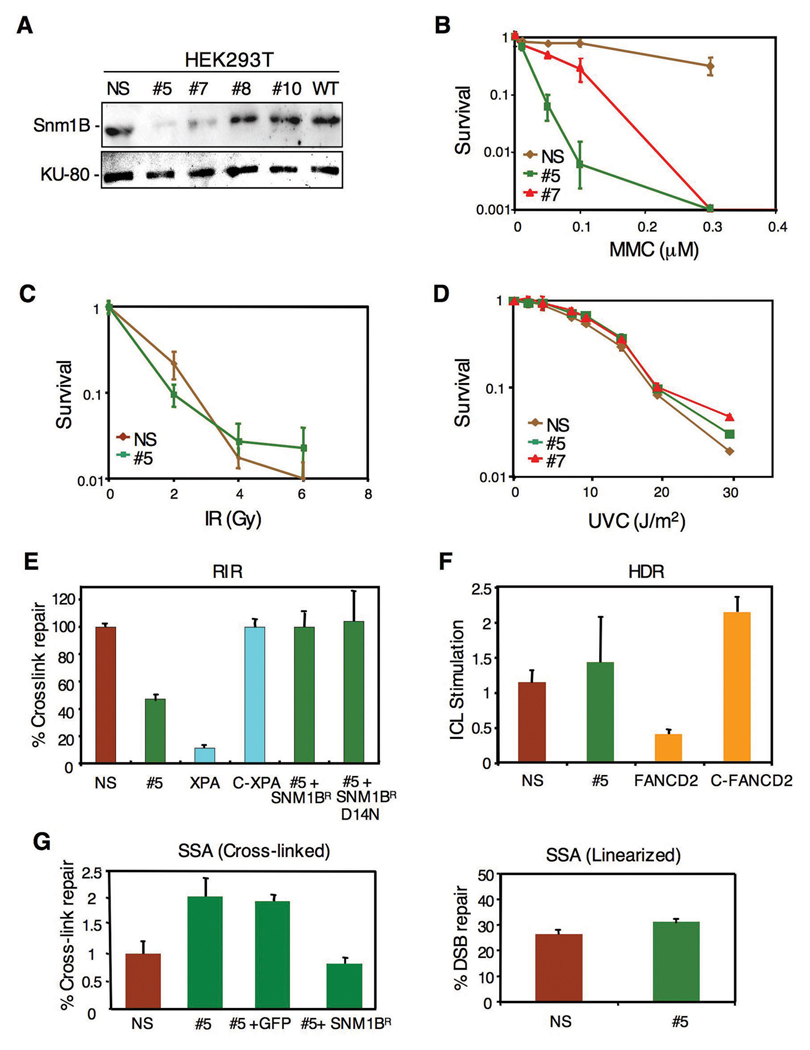
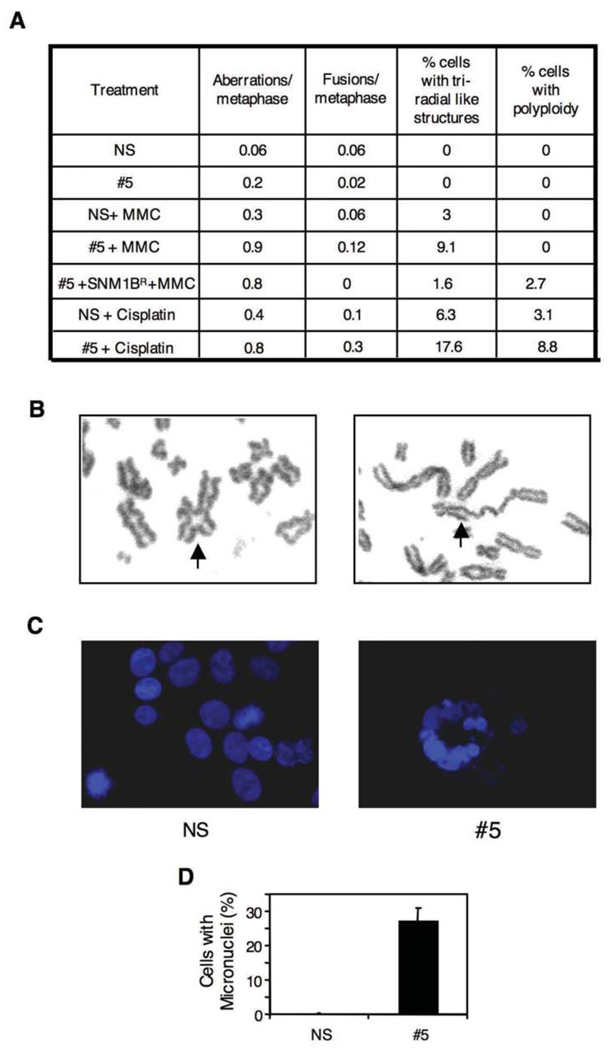
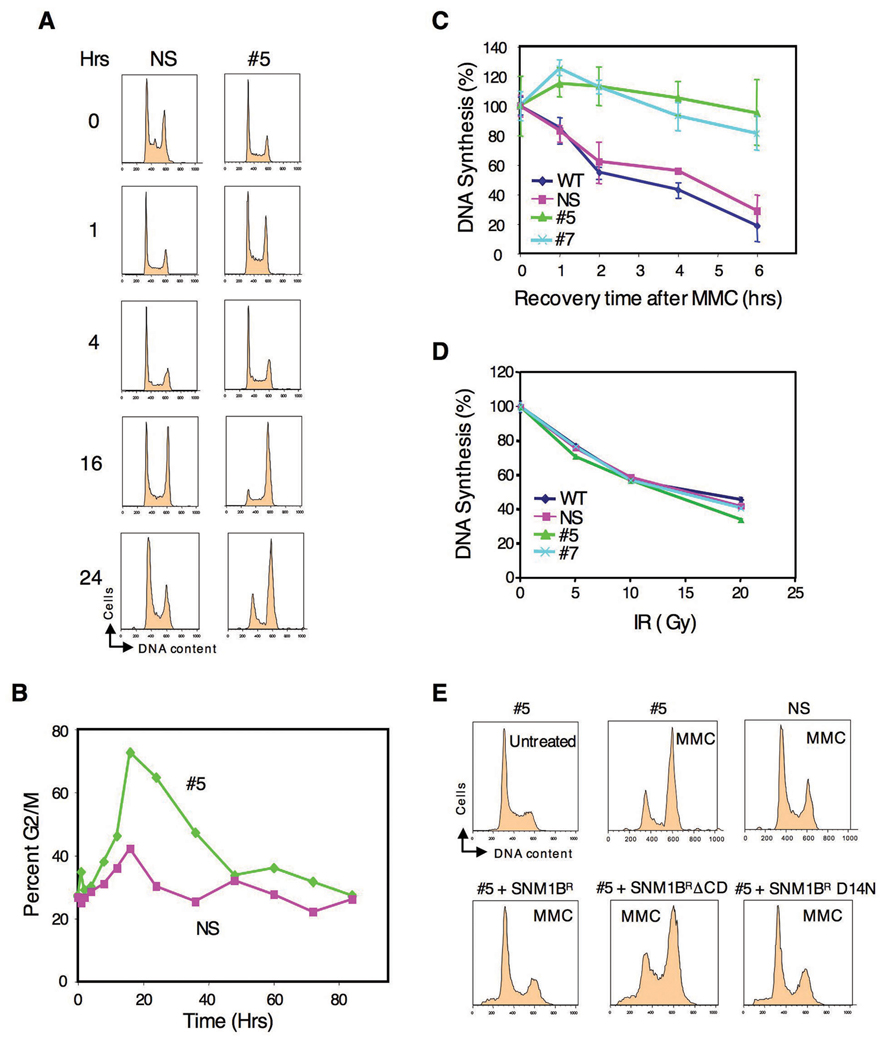
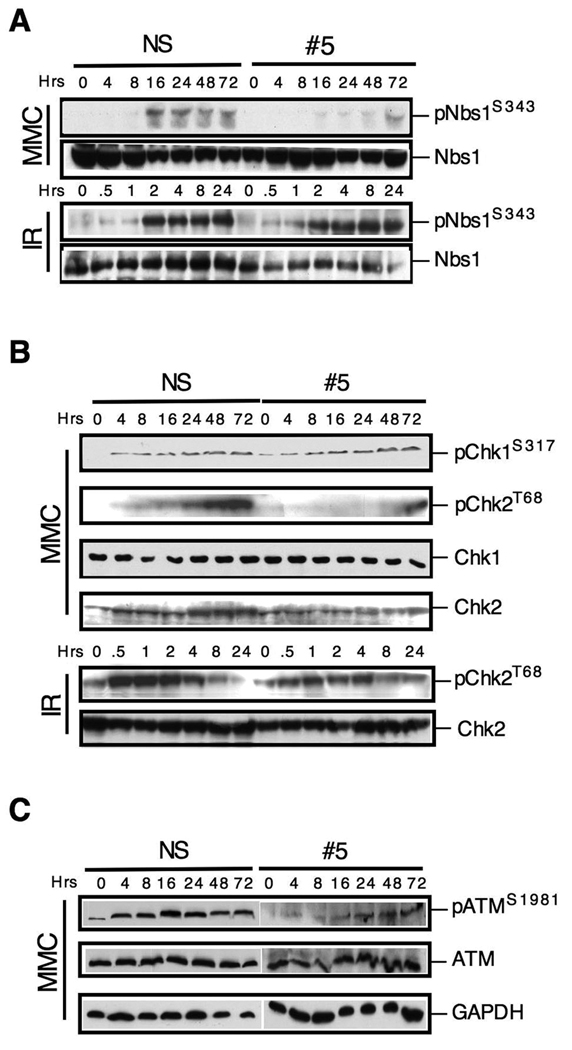
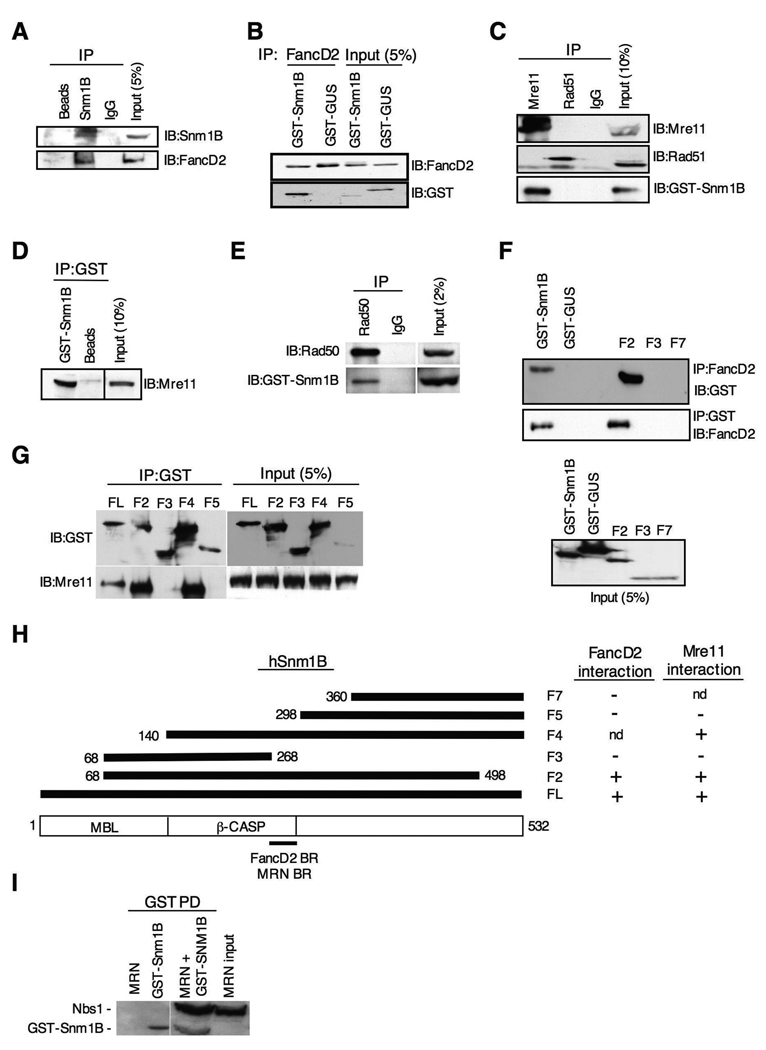
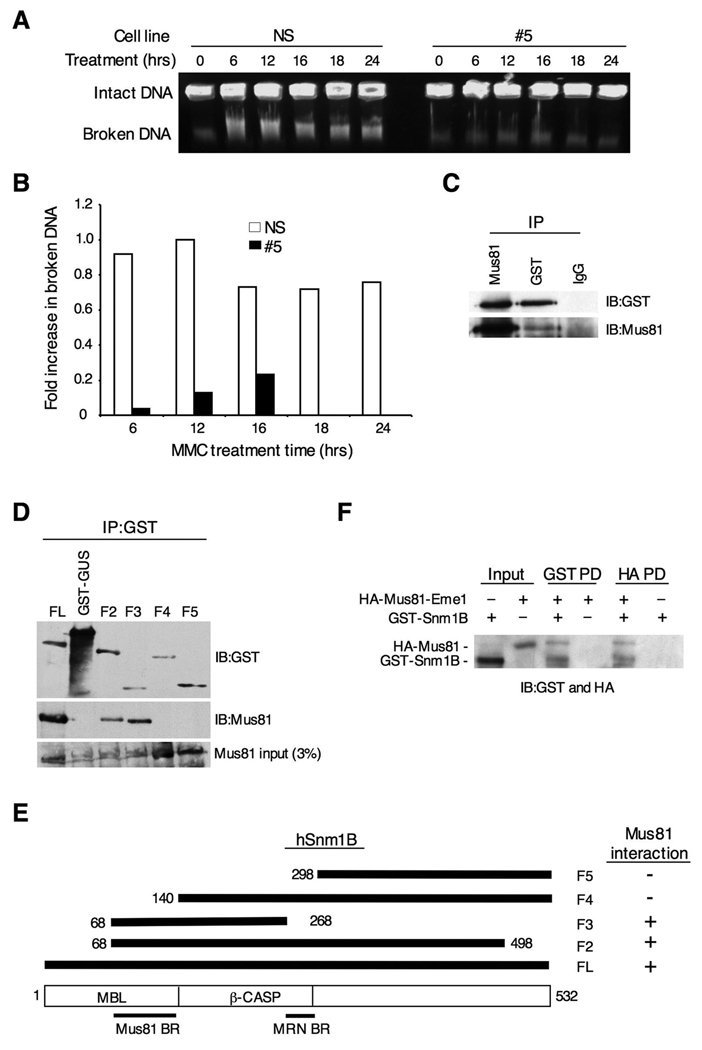
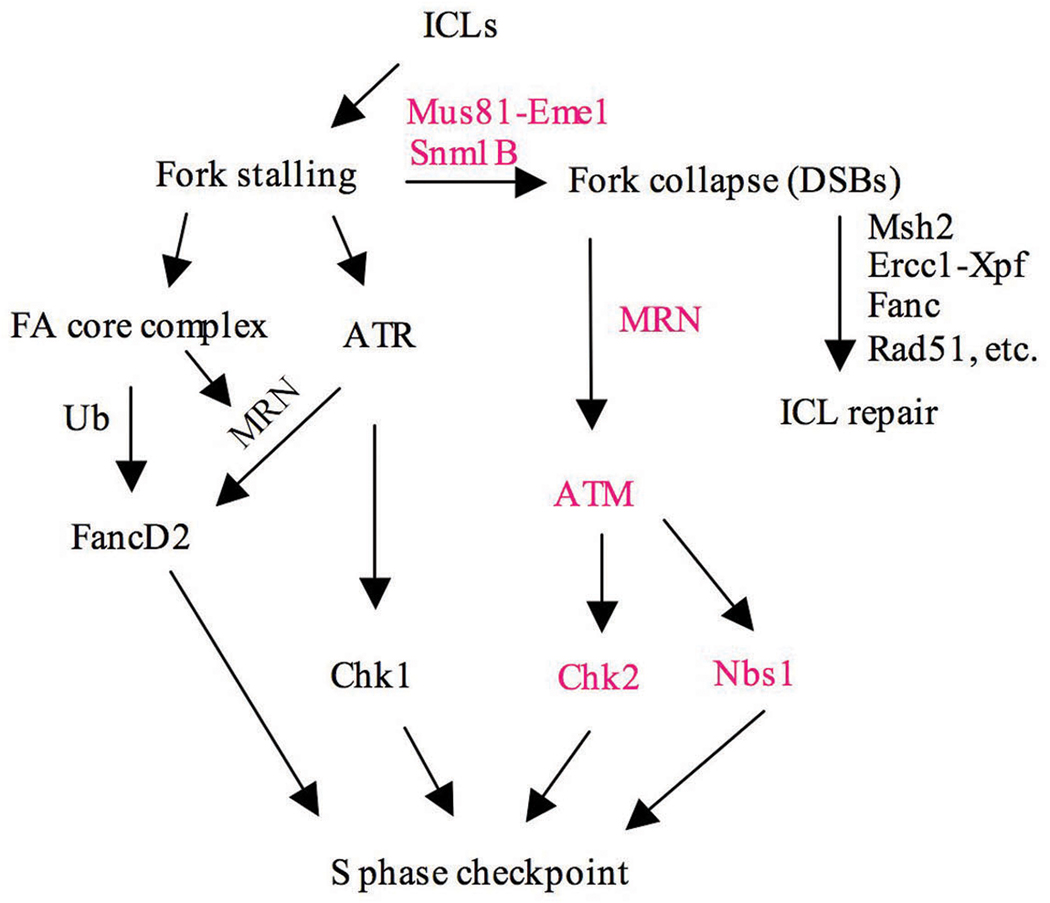
The removal of DNA interstrand cross-links (ICLs) has proven to be notoriously complicated due to the involvement of multiple pathways of DNA repair, which include the Fanconi anemia/BRCA pathway, homologous recombination and components of the nucleotide excision and mismatch repair pathways. Members of the SNM1 gene family have also been shown to have a role in mediating cellular resistance to ICLs, although their precise function has remained elusive. Here, we show that knockdown of Snm1B/Apollo in human cells results in hypersensitivity to mitomycin C (MMC), but not to IR. We also show that Snm1B-deficient cells exhibit a defective S phase checkpoint in response to MMC, but not to IR, and this finding may account for the specific sensitivity to the cross-linking drug. Interestingly, although previous studies have largely implicated ATR as the major kinase activated in response to ICLs, we show that it is activation of the ATM-mediated checkpoint that is defective in Snm1B-deficient cells. The requirement for Snm1B in ATM checkpoint activation specifically after ICL damage is correlated with its role in promoting double-strand break formation, and thus replication fork collapse. Consistent with this result Snm1B was found to interact directly with Mus81-Eme1, an endonuclease previously implicated in fork collapse. In addition, we also show that Snm1B interacts with the Mre11-Rad50-Nbs1 (MRN) complex and with FancD2 further substantiating its role as a checkpoint/DNA repair protein.
Figures
References
-
- Akkari YM, Bateman RL, Reifsteck CA, D'Andrea AD, Olson SB, Grompe M. The 4N cell cycle delay in Fanconi anemia reflects growth arrest in late S phase. Mol Genet Metab. 2001;74:403–412. - PubMed
-
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
