

A test on peptide stability of AMBER force fields with implicit solvation
- PMID: 18471007
- PMCID: PMC2699260
- DOI: 10.1021/jp800282x
A test on peptide stability of AMBER force fields with implicit solvation
Abstract
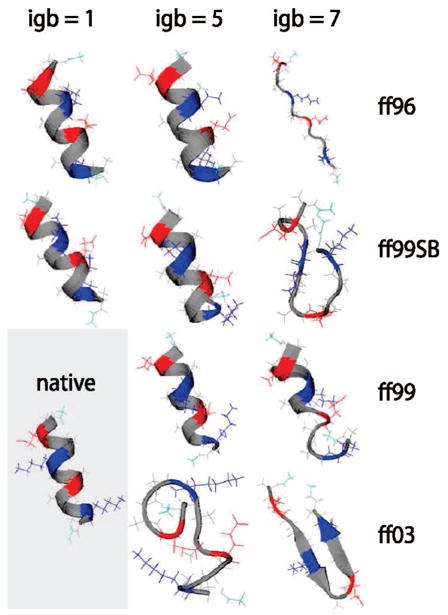
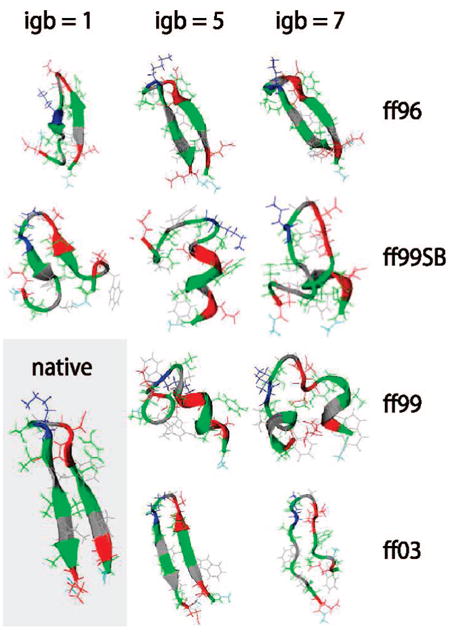
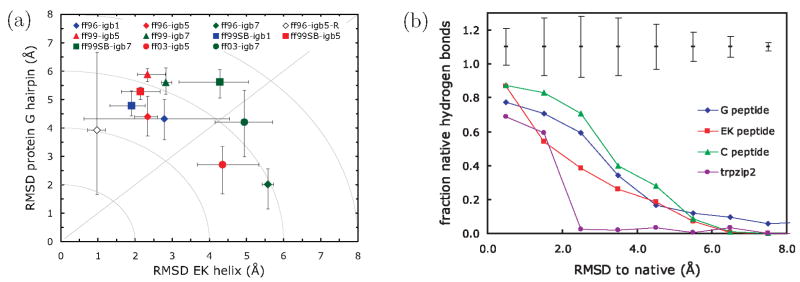
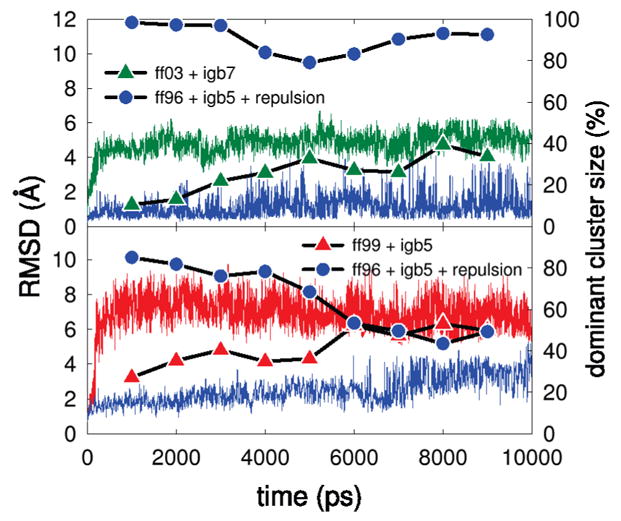
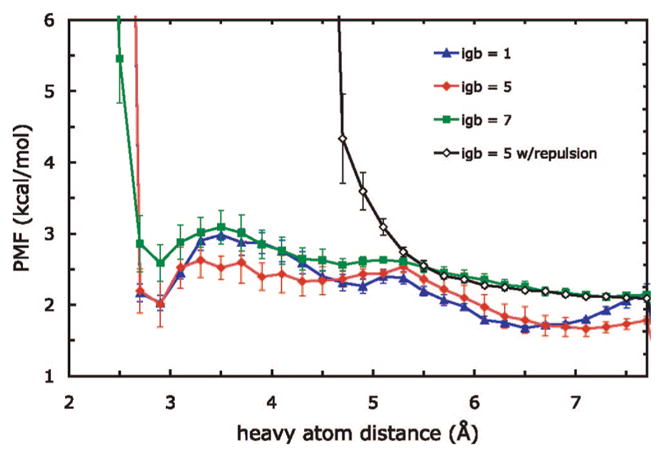
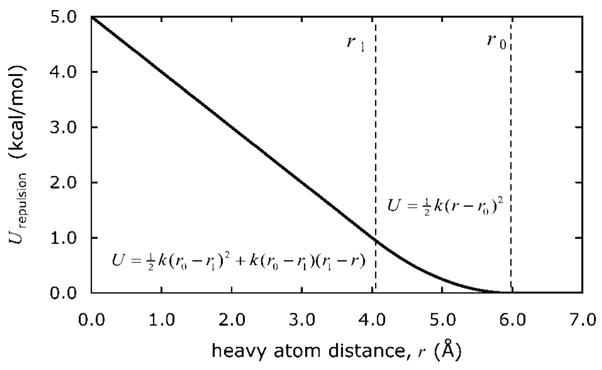
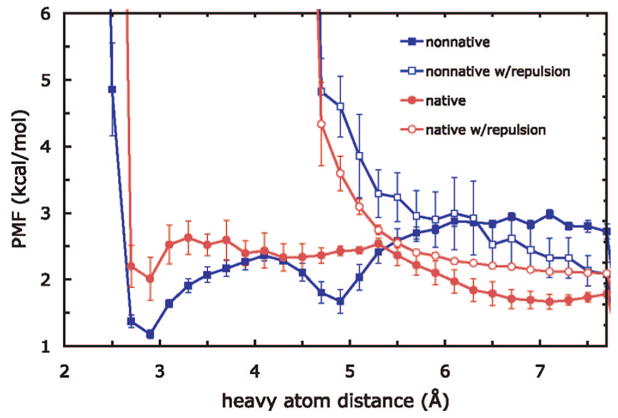
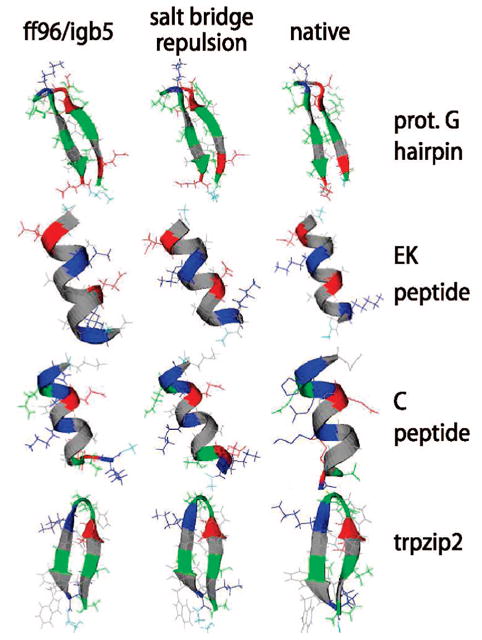
We used replica exchange molecular dynamics (REMD) simulations to evaluate four different AMBER force fields and three different implicit solvent models. Our aim was to determine if these physics-based models captured the correct secondary structures of two alpha-helical and two beta-peptides: the 14-mer EK helix of Baldwin and co-workers, the C-terminal helix of ribonuclease, the 16-mer C-terminal hairpin of protein G, and the trpzip2 miniprotein. The different models gave different results, but generally we found that AMBER ff96 plus the implicit solvent model of Onufriev, Bashford, and Case gave reasonable structures, and is fairly well-balanced between helix and sheet. We also observed differences in the strength of ion pairing in the solvent models, we but found that the native secondary structures were retained even when salt bridges were prevented in the conformational sampling. Overall, this work indicates that some of these all-atom physics-based force fields may be good starting points for protein folding and protein structure prediction.
Figures
References
-
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187.
-
- Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, III, Debolt S, Ferguson D, Seibel G, Kollman P. Comput Phys Commun. 1995;91:1.
-
- Jorgensen WL, Tirado-Rives J. J Am Chem Soc. 1988;110:1657. - PubMed
-
- Duan Y, Kollman PA. Science. 1998;282:740. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
