

P53, cyclin-dependent kinase and abnormal amplification of centrosomes
- PMID: 18472015
- PMCID: PMC2647860
- DOI: 10.1016/j.bbcan.2008.04.002
P53, cyclin-dependent kinase and abnormal amplification of centrosomes
Abstract
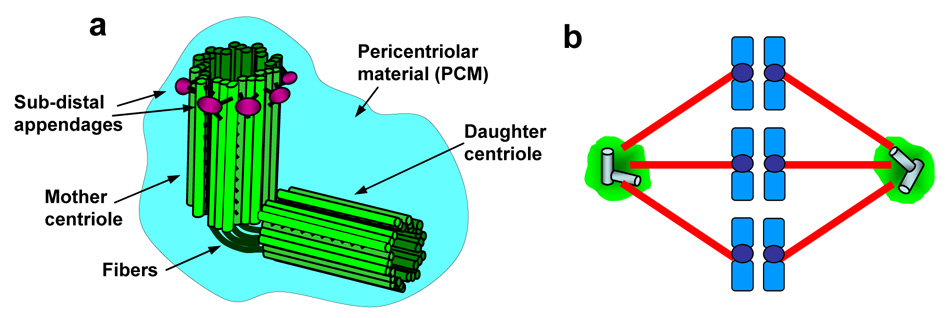
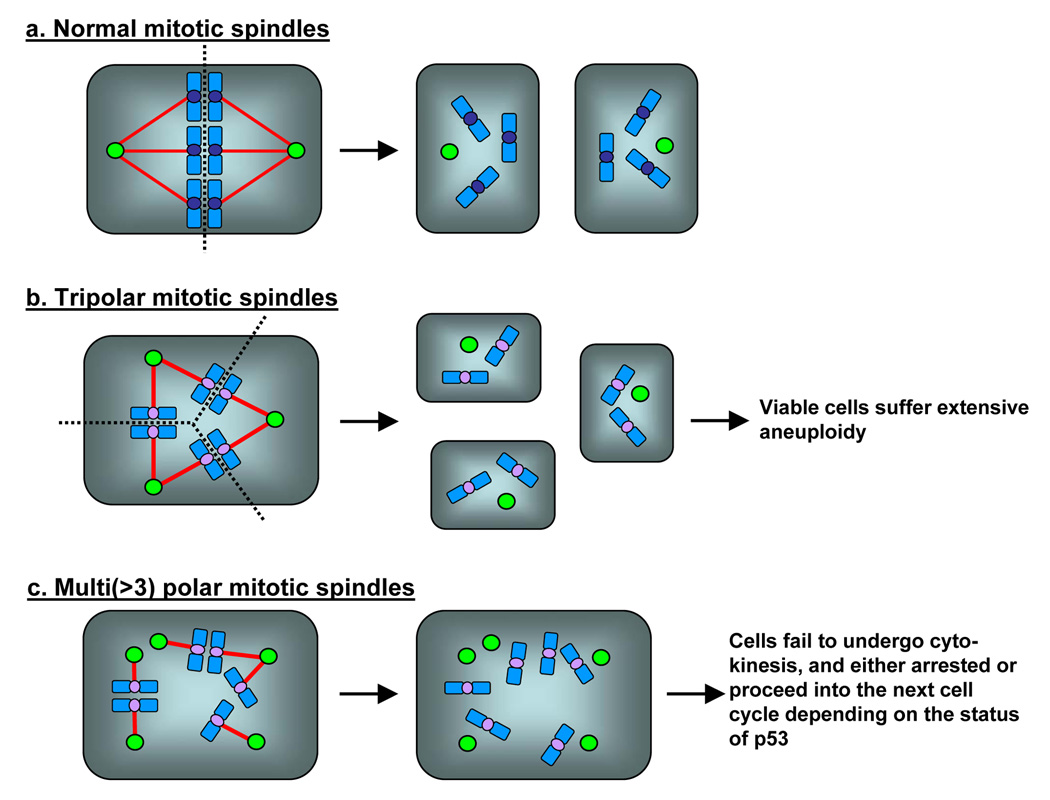
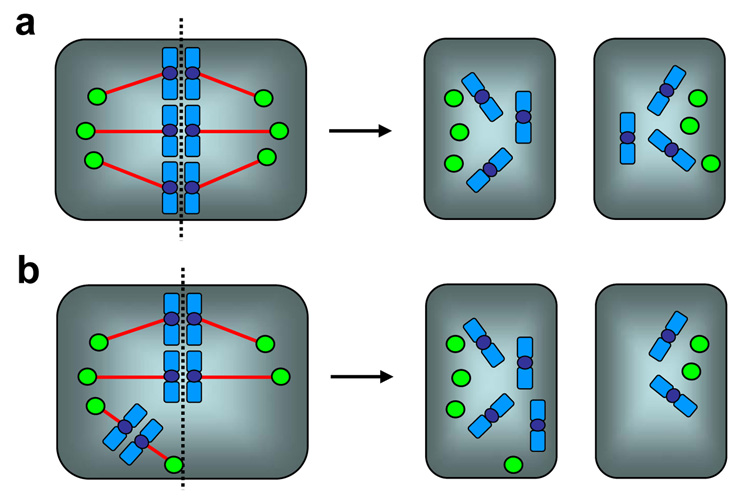
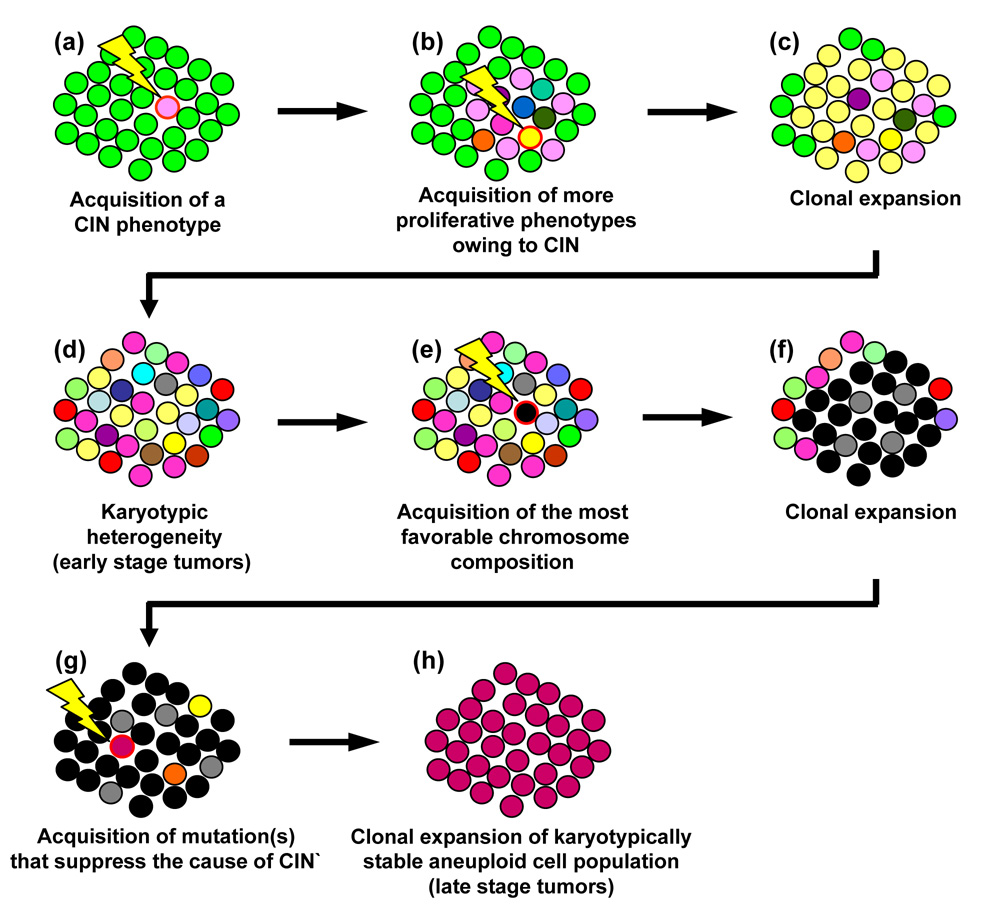
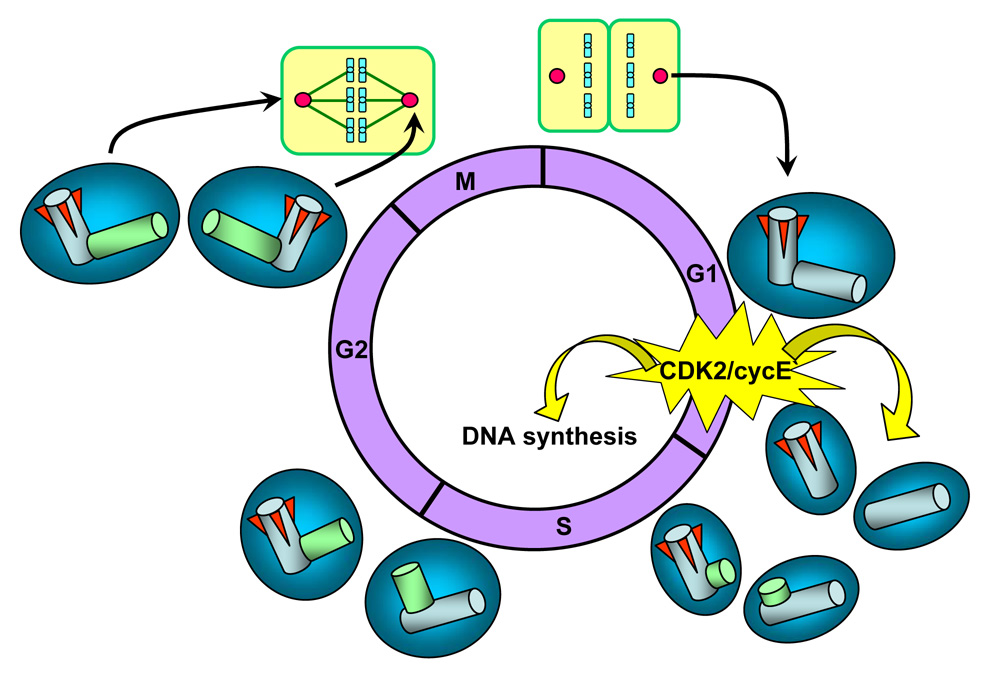
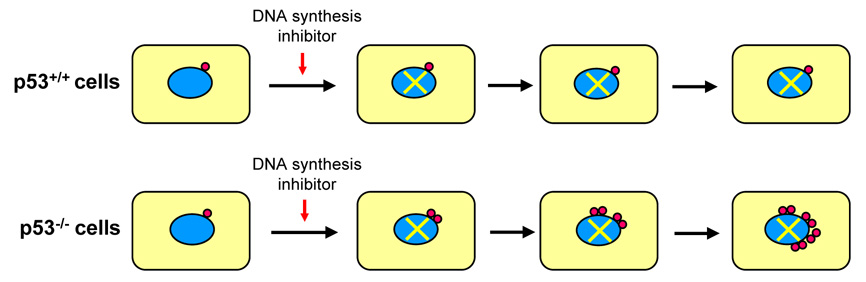
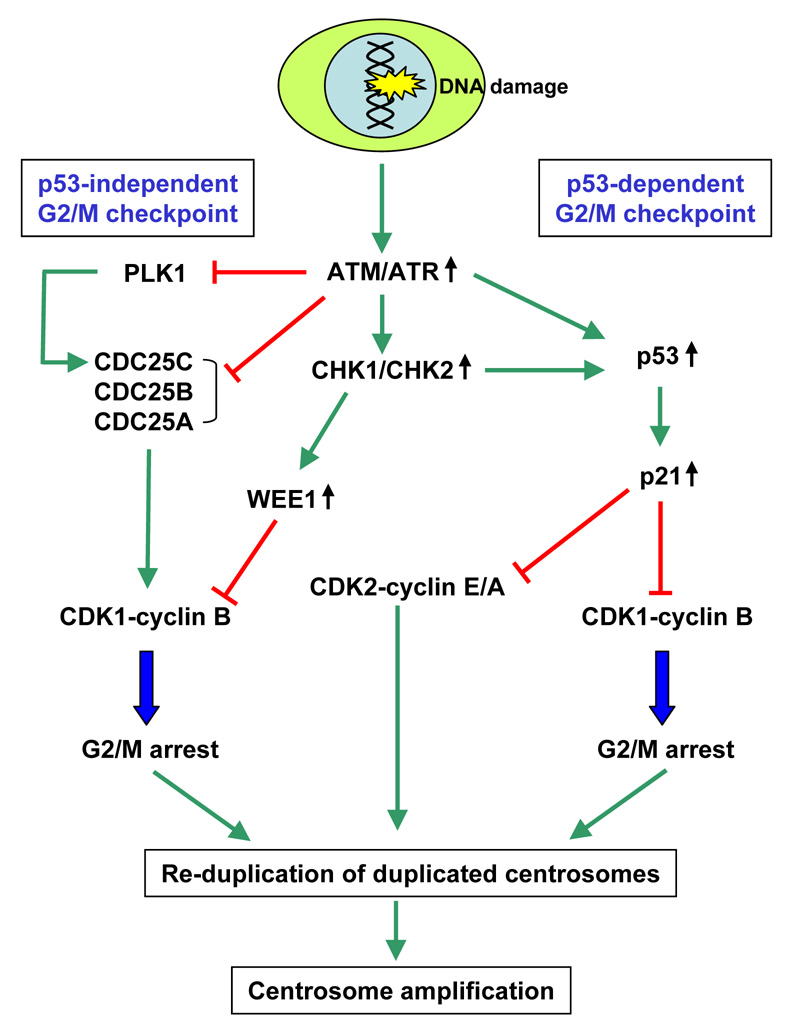
Centrosomes play a critical role in formation of bipolar mitotic spindles, an essential event for accurate chromosome segregation into daughter cells. Numeral abnormalities of centrosomes (centrosome amplification) occur frequently in cancers, and are considered to be the major cause of chromosome instability, which accelerates acquisition of malignant phenotypes during tumor progression. Loss or mutational inactivation of p53 tumor suppressor protein, one of the most common mutations found in cancers, results in a high frequency of centrosome amplification in part via allowing the activation of the cyclin-dependent kinase (CDK) 2-cyclin E (as well as CDK2-cyclin A) which is a key factor for the initiation of centrosome duplication. In this review, the role of centrosome amplification in tumor progression, and mechanistic view of how centrosomes are amplified in cells through focusing on loss of p53 and aberrant activities of CDK2-cyclins will be discussed.
Figures
Similar articles
-
Roles of cyclins A and E in induction of centrosome amplification in p53-compromised cells.Oncogene. 2008 Sep 11;27(40):5288-302. doi: 10.1038/onc.2008.161. Epub 2008 May 19. Oncogene. 2008. PMID: 18490919 Free PMC article.
-
Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression.Oncogene. 2000 Mar 23;19(13):1635-46. doi: 10.1038/sj.onc.1203460. Oncogene. 2000. PMID: 10763820
-
Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway.Oncogene. 2001 May 31;20(25):3173-84. doi: 10.1038/sj.onc.1204424. Oncogene. 2001. PMID: 11423967
-
Centrosome amplification, chromosome instability and cancer development.Cancer Lett. 2005 Dec 8;230(1):6-19. doi: 10.1016/j.canlet.2004.12.028. Cancer Lett. 2005. PMID: 16253756 Review.
-
Oncogenes and tumour suppressors take on centrosomes.Nat Rev Cancer. 2007 Dec;7(12):911-24. doi: 10.1038/nrc2249. Nat Rev Cancer. 2007. PMID: 18004399 Review.
Cited by
-
Porcine circovirus type 2 ORF3 protein induces apoptosis in melanoma cells.BMC Cancer. 2018 Dec 10;18(1):1237. doi: 10.1186/s12885-018-5090-2. BMC Cancer. 2018. PMID: 30526524 Free PMC article.
-
Centrosome Aberration Frequency and Disease Association in B-Acute Lymphoblastic Leukemia.In Vivo. 2017 Mar-Apr;31(2):215-220. doi: 10.21873/invivo.11048. In Vivo. 2017. PMID: 28358703 Free PMC article.
-
Cdk4 and nek2 signal binucleation and centrosome amplification in a her2+ breast cancer model.PLoS One. 2013 Jun 11;8(6):e65971. doi: 10.1371/journal.pone.0065971. Print 2013. PLoS One. 2013. PMID: 23776583 Free PMC article.
-
Specific overexpression of cyclin E·CDK2 in early preinvasive and primary breast tumors in female ACI rats induced by estrogen.Horm Cancer. 2010 Feb;1(1):34-43. doi: 10.1007/s12672-009-0004-z. Epub 2010 Feb 10. Horm Cancer. 2010. PMID: 21761349 Free PMC article.
-
Cdk2 and Cdk4 regulate the centrosome cycle and are critical mediators of centrosome amplification in p53-null cells.Mol Cell Biol. 2010 Feb;30(3):694-710. doi: 10.1128/MCB.00253-09. Epub 2009 Nov 23. Mol Cell Biol. 2010. PMID: 19933848 Free PMC article.
References
-
- Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr. Opin. Cell Biol. 2002;14:25–34. - PubMed
-
- Sattler CA, Sawada N, Sattler GL, Pitot HC. Electron microscopic and time lapse studies of mitosis in cultured rat hepatocytes. Hepatology. 1988;8:1540–1549. - PubMed
-
- Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230:6–19. - PubMed
-
- Levine DS, Sanchez CA, Rabinovitch PS, Reid BJ. Formation of the tetraploid intermediate is associated with the development of cells with more than four centrioles in the elastase-simian virus 40 tumor antigen transgenic mouse model of pancreatic cancer. Proc. Natl. Acad. Sci. USA. 1991;88:6427–6431. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
