

Polynucleotide kinase as a potential target for enhancing cytotoxicity by ionizing radiation and topoisomerase I inhibitors
- PMID: 18473721
- PMCID: PMC2962422
- DOI: 10.2174/187152008784220311
Polynucleotide kinase as a potential target for enhancing cytotoxicity by ionizing radiation and topoisomerase I inhibitors
Abstract
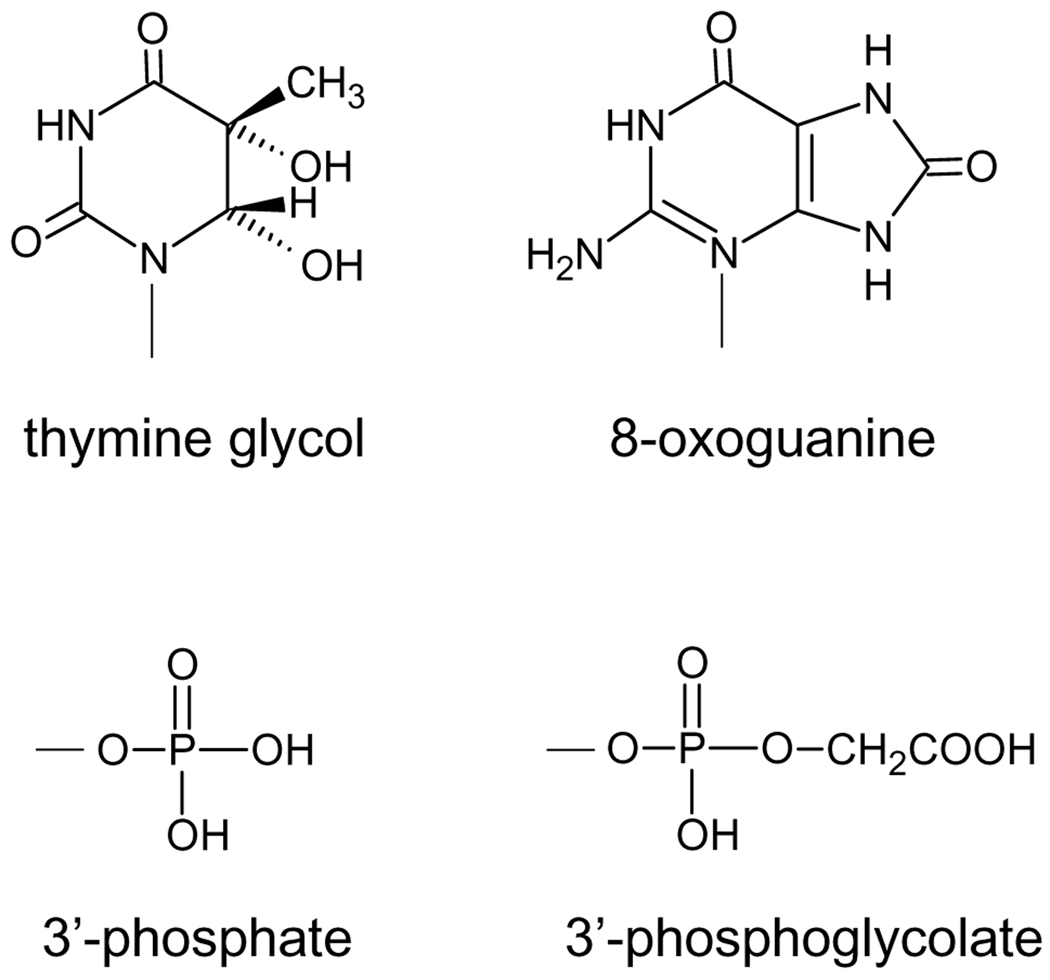
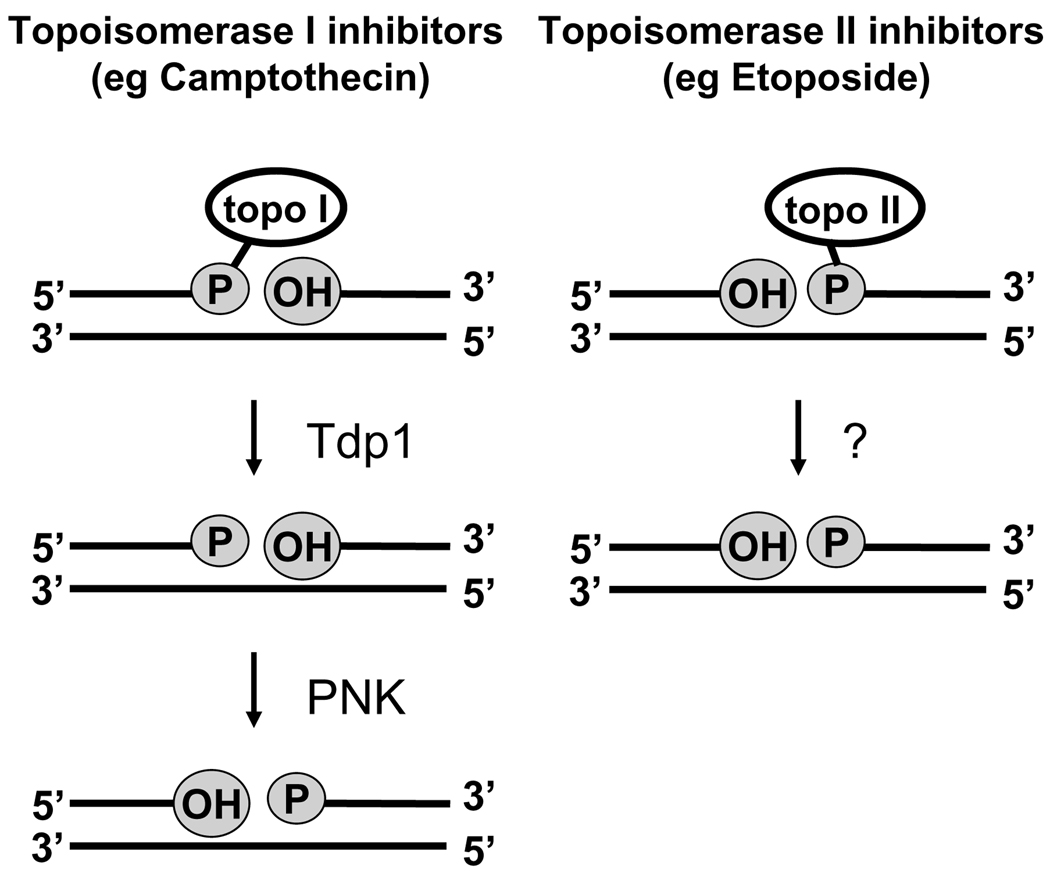
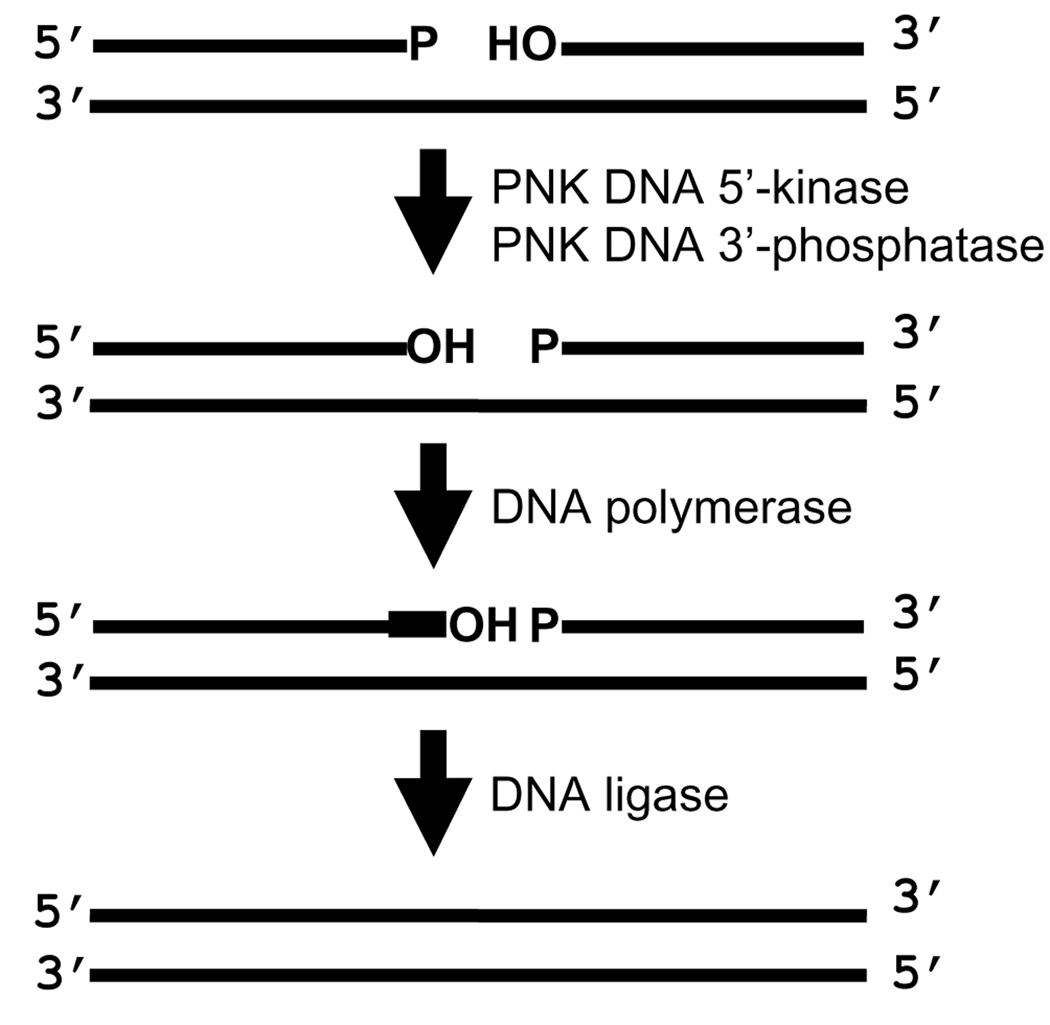
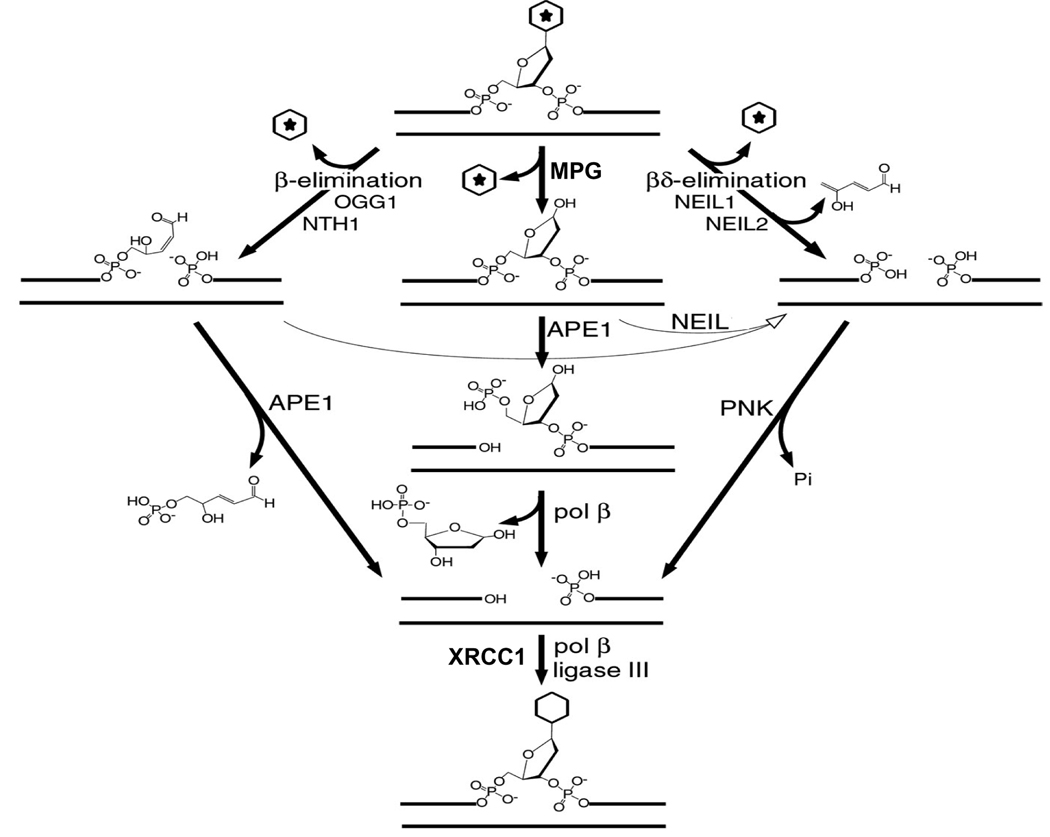
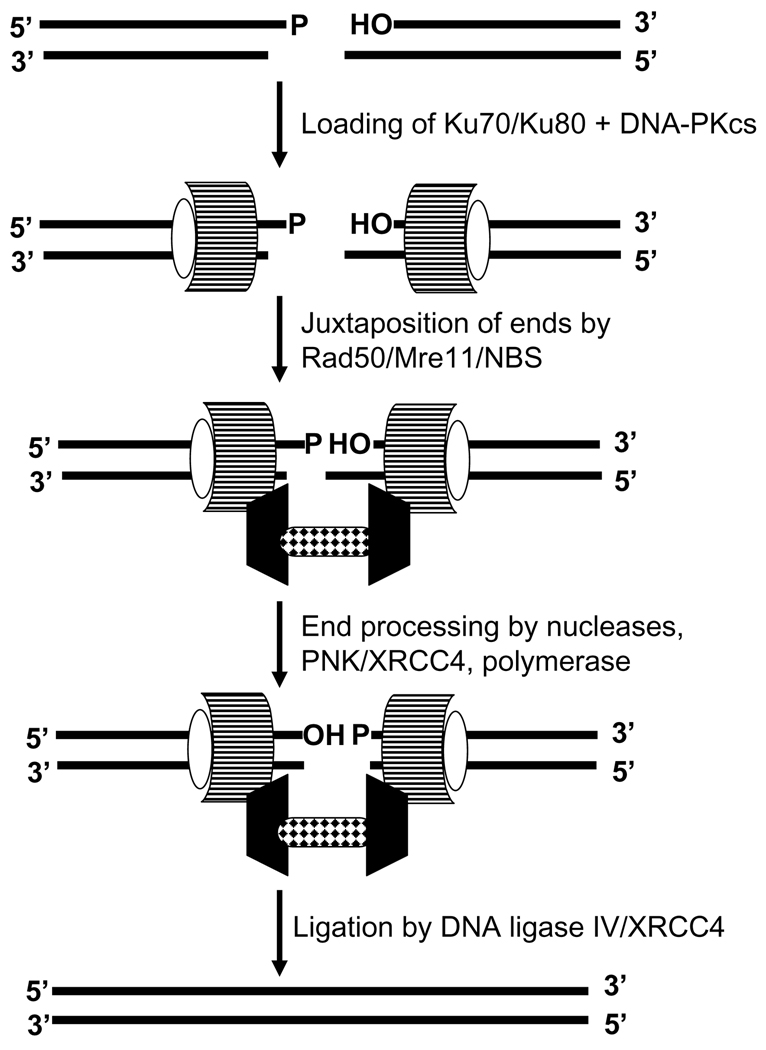
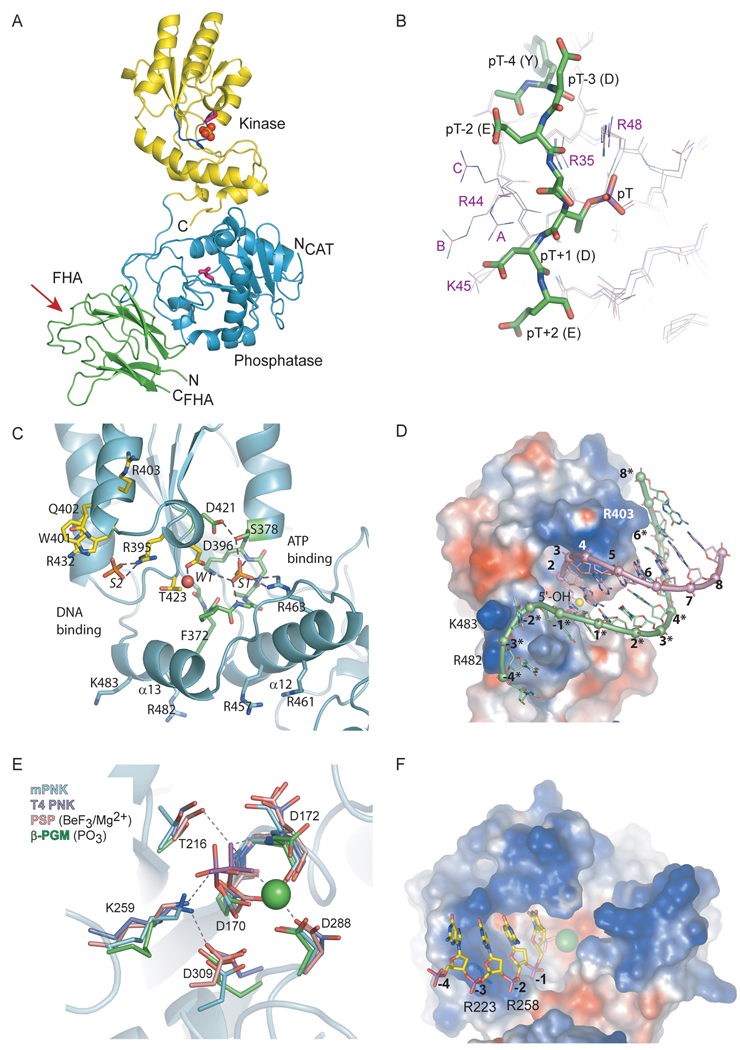
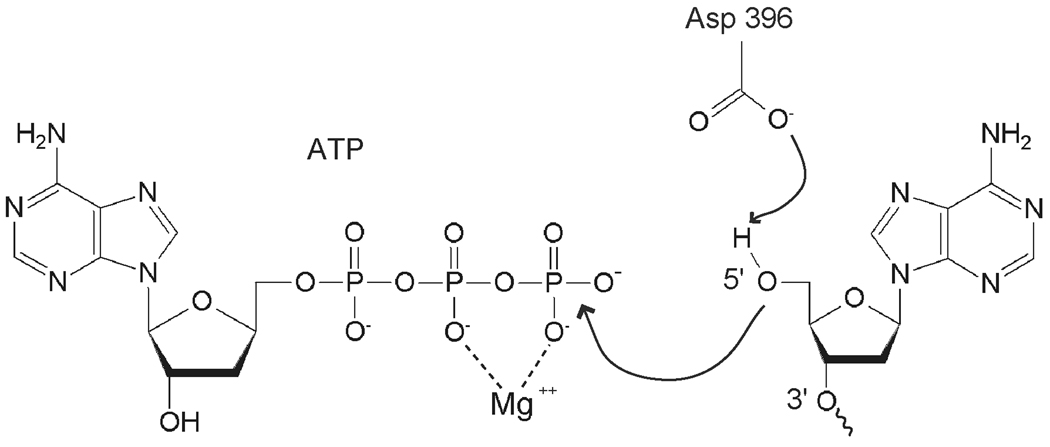
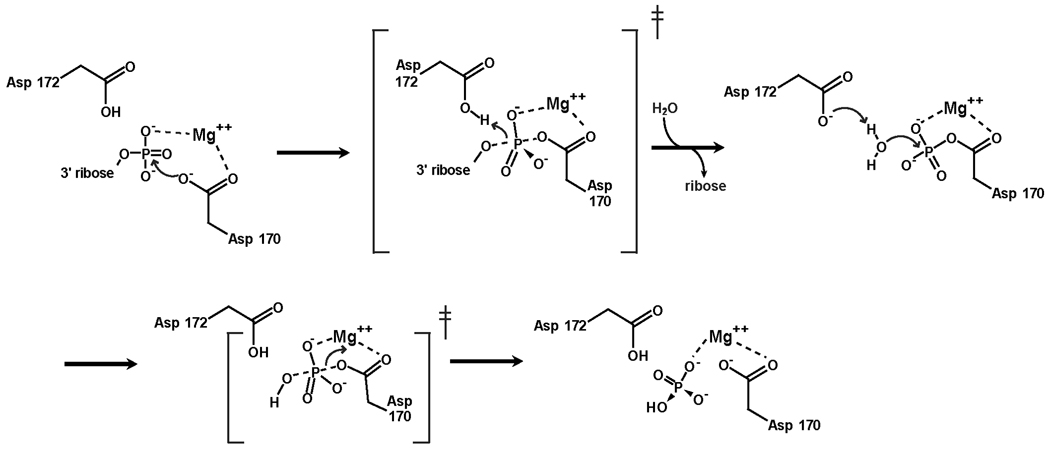
The cytotoxicity of many antineoplastic agents is due to their capacity to damage DNA and there is evidence indicating that DNA repair contributes to the cellular resistance to such agents. DNA strand breaks constitute a significant proportion of the lesions generated by a broad range of genotoxic agents, either directly, or during the course of DNA repair. Strand breaks that are caused by many agents including ionizing radiation, topoisomerase I inhibitors, and DNA repair glycosylases such as NEIL1 and NEIL2, often contain 5'-hydroxyl and/or 3'-phosphate termini. These ends must be converted to 5'-phosphate and 3'-hydroxyl termini in order to allow DNA polymerases and ligases to catalyze repair synthesis and strand rejoining. A key enzyme involved in this end-processing is polynucleotide kinase (PNK), which possesses two enzyme activities, a DNA 5'-kinase activity and a 3'-phosphatase activity. PNK participates in the single-strand break repair pathway and the non-homologous end joining pathway for double-strand break repair. RNAi-mediated down-regulation of PNK renders cells more sensitive to ionizing radiation and camptothecin, a topoisomerase I inhibitor. Structural analysis of PNK revealed the protein is composed of three domains, the kinase domain at the C-terminus, the phosphatase domain in the centre and a forkhead associated (FHA) domain at the N-terminus. The FHA domain plays a critical role in the binding of PNK to other DNA repair proteins. Thus each PNK domain may be a suitable target for small molecule inhibition to effectively reduce resistance to ionizing radiation and topoisomerase I inhibitors.
Figures
Similar articles
-
DNA end-processing enzyme polynucleotide kinase as a potential target in the treatment of cancer.Future Oncol. 2010 Jun;6(6):1031-42. doi: 10.2217/fon.10.40. Future Oncol. 2010. PMID: 20528239
-
Deinococcus radiodurans HD-Pnk, a Nucleic Acid End-Healing Enzyme, Abets Resistance to Killing by Ionizing Radiation and Mitomycin C.J Bacteriol. 2018 Aug 10;200(17):e00151-18. doi: 10.1128/JB.00151-18. Print 2018 Sep 1. J Bacteriol. 2018. PMID: 29891641 Free PMC article.
-
The phosphatase activity of mammalian polynucleotide kinase takes precedence over its kinase activity in repair of single strand breaks.Nucleic Acids Res. 2006 Apr 28;34(8):2230-7. doi: 10.1093/nar/gkl275. Print 2006. Nucleic Acids Res. 2006. PMID: 16648365 Free PMC article.
-
Inhibition of Topoisomerase (DNA) I (TOP1): DNA Damage Repair and Anticancer Therapy.Biomolecules. 2015 Jul 22;5(3):1652-70. doi: 10.3390/biom5031652. Biomolecules. 2015. PMID: 26287259 Free PMC article. Review.
-
Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells.Cell Res. 2008 Jan;18(1):27-47. doi: 10.1038/cr.2008.8. Cell Res. 2008. PMID: 18166975 Free PMC article. Review.
Cited by
-
Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair.Trends Biochem Sci. 2011 May;36(5):262-71. doi: 10.1016/j.tibs.2011.01.006. Epub 2011 Feb 25. Trends Biochem Sci. 2011. PMID: 21353781 Free PMC article. Review.
-
Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage.Nucleic Acids Res. 2011 Nov;39(21):9224-37. doi: 10.1093/nar/gkr647. Epub 2011 Aug 8. Nucleic Acids Res. 2011. PMID: 21824916 Free PMC article.
-
Hitting the bull's eye: novel directed cancer therapy through helicase-targeted synthetic lethality.J Cell Biochem. 2009 Apr 1;106(5):758-63. doi: 10.1002/jcb.22048. J Cell Biochem. 2009. PMID: 19173305 Free PMC article. Review.
-
Nano-Delivery of a Novel Inhibitor of Polynucleotide Kinase/Phosphatase (PNKP) for Targeted Sensitization of Colorectal Cancer to Radiation-Induced DNA Damage.Front Oncol. 2021 Dec 23;11:772920. doi: 10.3389/fonc.2021.772920. eCollection 2021. Front Oncol. 2021. PMID: 35004293 Free PMC article.
-
Gold(III) macrocycles: nucleotide-specific unconventional catalytic inhibitors of human topoisomerase I.J Am Chem Soc. 2014 Apr 16;136(15):5670-82. doi: 10.1021/ja412350f. Epub 2014 Apr 2. J Am Chem Soc. 2014. PMID: 24694294 Free PMC article.
References
-
- Gatti L, Zunino F. Methods Mol. Med. 2005;111:127. - PubMed
-
- Madhusudan S, Middleton MR. Cancer Treat. Rev. 2005;31:603. - PubMed
-
- Murray D, Begg AC. In: In DNA Repair in Cancer Therapy. Panasci LC, Alaoui-Jamali MA, editors. Totowa: Humana Press; 2004. pp. 211–256.
-
- Ding J, Miao ZH, Meng LH, Geng MY. Trends Pharmacol. Sci. 2006;27:338. - PubMed
-
- Sanchez-Perez I. Clin. Transl. Oncol. 2006;8:642. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
