

Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue
- PMID: 18480843
- PMCID: PMC2426729
- DOI: 10.1038/emboj.2008.92
Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue
Abstract
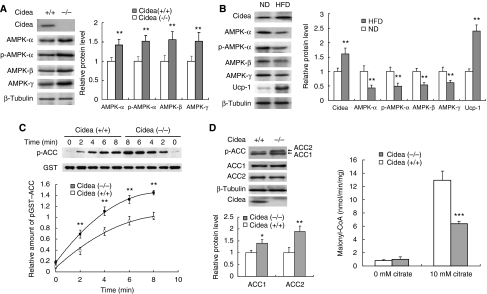
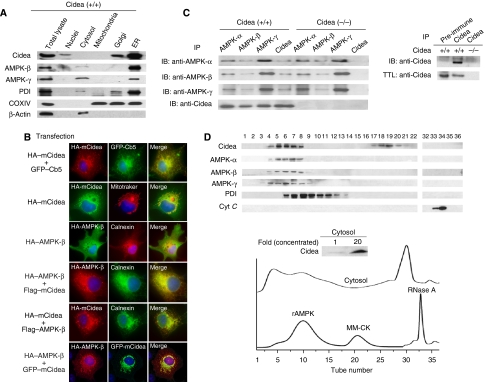
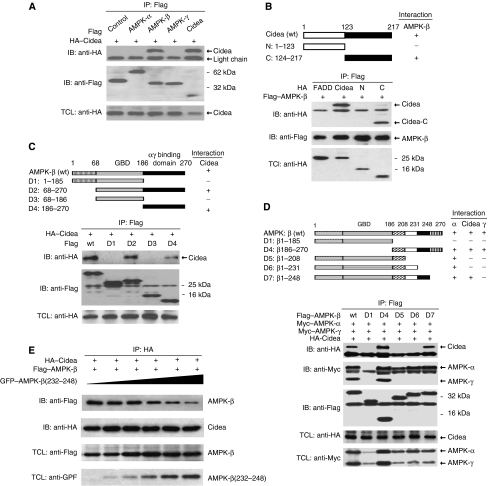
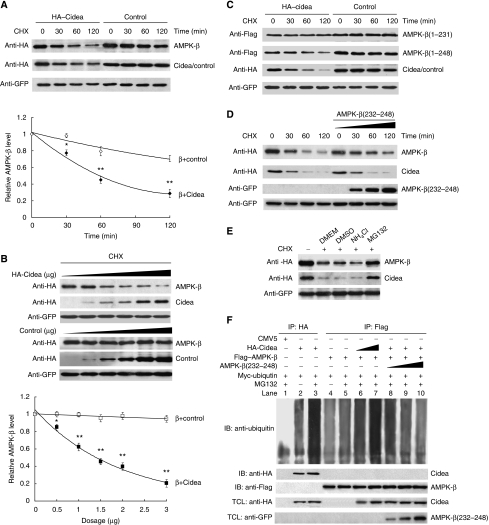
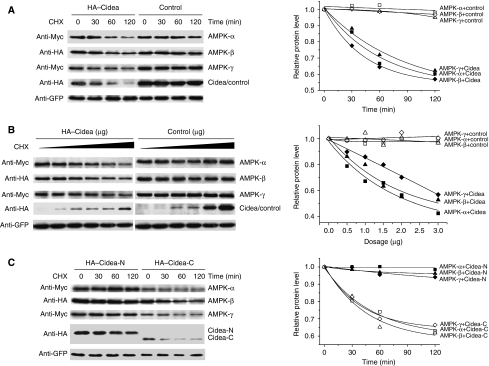
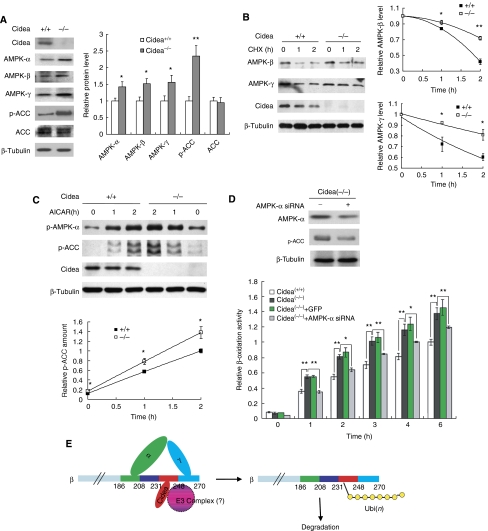
We previously showed that Cidea(-/-) mice are resistant to diet-induced obesity through the upregulation of energy expenditure. The AMP-activated protein kinase (AMPK), consisting of catalytic alpha subunit and regulatory subunits beta and gamma, has a pivotal function in energy homoeostasis. We show here that AMPK protein levels and enzymatic activity were significantly increased in the brown adipose tissue of Cidea(-/-) mice. We also found that Cidea is colocalized with AMPK in the endoplasmic reticulum and forms a complex with AMPK in vivo through specific interaction with the beta subunit of AMPK, but not with the alpha or gamma subunit. When co-expressed with Cidea, the stability of AMPK-beta subunit was dramatically reduced due to increased ubiquitination-mediated degradation, which depends on a physical interaction between Cidea and AMPK. Furthermore, AMPK stability and enzymatic activity were increased in Cidea(-/-) adipocytes differentiated from mouse embryonic fibroblasts or preadipocytes. Our data strongly suggest that AMPK can be regulated by Cidea-mediated ubiquitin-dependent proteosome degradation, and provide a molecular explanation for the increased energy expenditure and lean phenotype in Cidea-null mice.
Figures
References
-
- Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, Esposito D, Gillette WK, Hopkins RF III, Hartley JL, Furihata M, Oishi S, Zhen W, Burke TR Jr, Linehan WM, Schmidt LS, Zbar B (2006) Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA 103: 15552–15557 - PMC - PubMed
-
- Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, Steppan CM, Ahima RS, Obici S, Rossetti L, Lazar MA (2004) Regulation of fasted blood glucose by resistin. Science 303: 1195–1198 - PubMed
-
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG et al. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444: 337–342 - PMC - PubMed
-
- Chen Z, Guo K, Toh SY, Zhou Z, Li P (2000) Mitochondria localization and dimerization are required for CIDE-B to induce apoptosis. J Biol Chem 275: 22619–22622 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
