

SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase
- PMID: 18482975
- PMCID: PMC2459285
- DOI: 10.1074/jbc.M802187200
SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase
Abstract
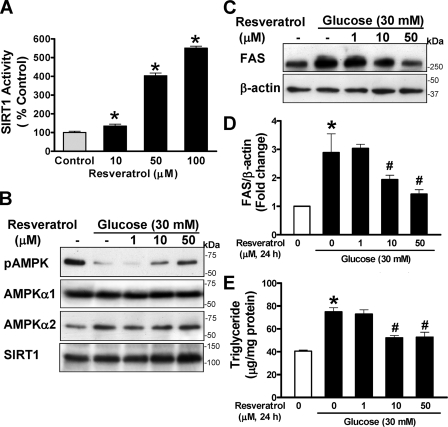
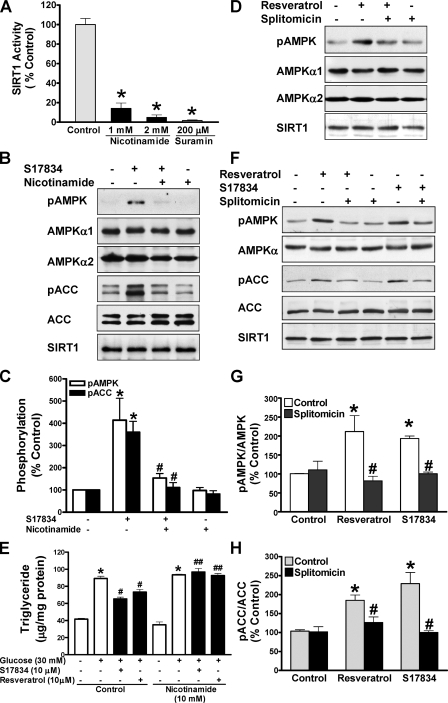
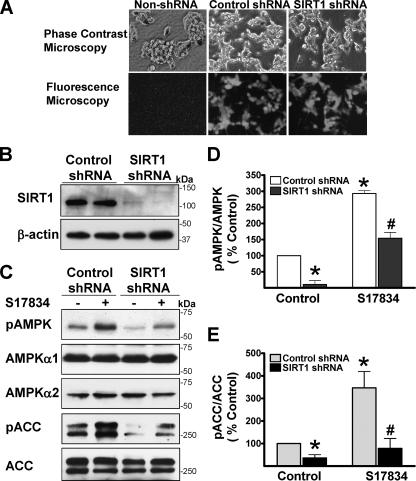
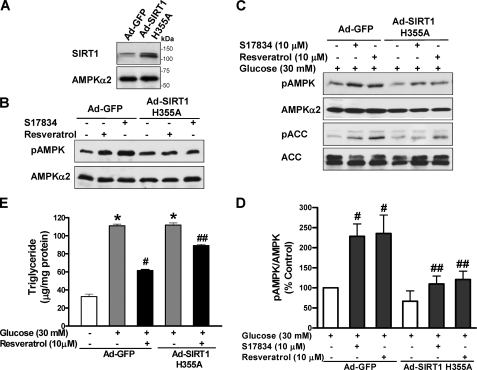
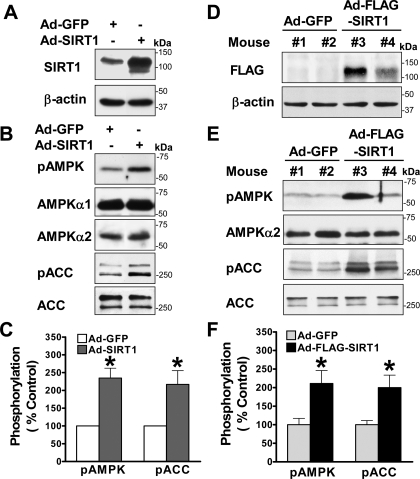
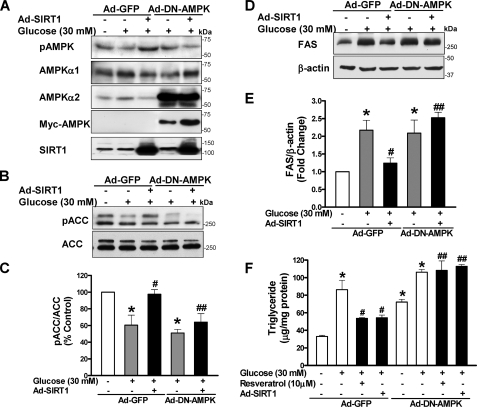
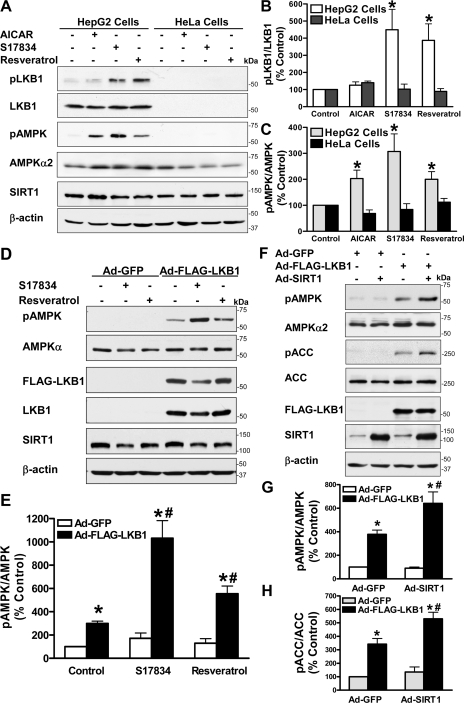
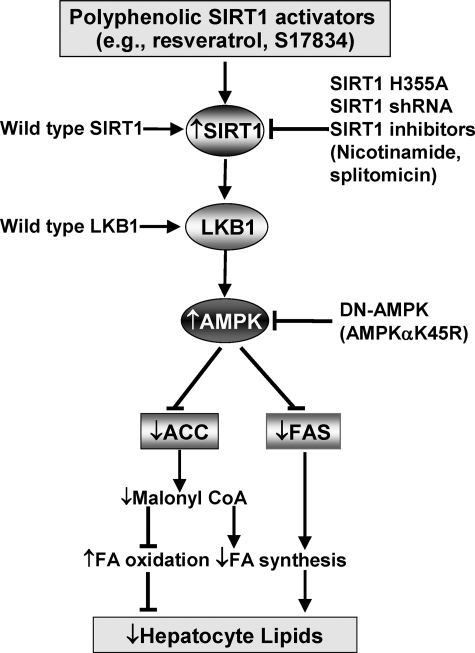
Resveratrol may protect against metabolic disease through activating SIRT1 deacetylase. Because we have recently defined AMPK activation as a key mechanism for the beneficial effects of polyphenols on hepatic lipid accumulation, hyperlipidemia, and atherosclerosis in type 1 diabetic mice, we hypothesize that polyphenol-activated SIRT1 acts upstream of AMPK signaling and hepatocellular lipid metabolism. Here we show that polyphenols, including resveratrol and the synthetic polyphenol S17834, increase SIRT1 deacetylase activity, LKB1 phosphorylation at Ser(428), and AMPK activity. Polyphenols substantially prevent the impairment in phosphorylation of AMPK and its downstream target, ACC (acetyl-CoA carboxylase), elevation in expression of FAS (fatty acid synthase), and lipid accumulation in human HepG2 hepatocytes exposed to high glucose. These effects of polyphenols are largely abolished by pharmacological and genetic inhibition of SIRT1, suggesting that the stimulation of AMPK and lipid-lowering effect of polyphenols depend on SIRT1 activity. Furthermore, adenoviral overexpression of SIRT1 stimulates the basal AMPK signaling in HepG2 cells and in the mouse liver. AMPK activation by SIRT1 also protects against FAS induction and lipid accumulation caused by high glucose. Moreover, LKB1, but not CaMKKbeta, is required for activation of AMPK by polyphenols and SIRT1. These findings suggest that SIRT1 functions as a novel upstream regulator for LKB1/AMPK signaling and plays an essential role in the regulation of hepatocyte lipid metabolism. Targeting SIRT1/LKB1/AMPK signaling by polyphenols may have potential therapeutic implications for dyslipidemia and accelerated atherosclerosis in diabetes and age-related diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
