

Mutations in the MESP2 gene cause spondylothoracic dysostosis/Jarcho-Levin syndrome
- PMID: 18485326
- PMCID: PMC2427230
- DOI: 10.1016/j.ajhg.2008.04.014
Mutations in the MESP2 gene cause spondylothoracic dysostosis/Jarcho-Levin syndrome
Abstract
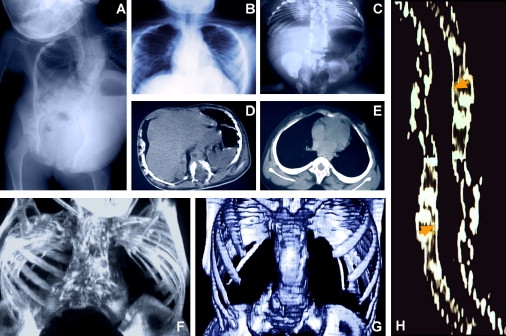
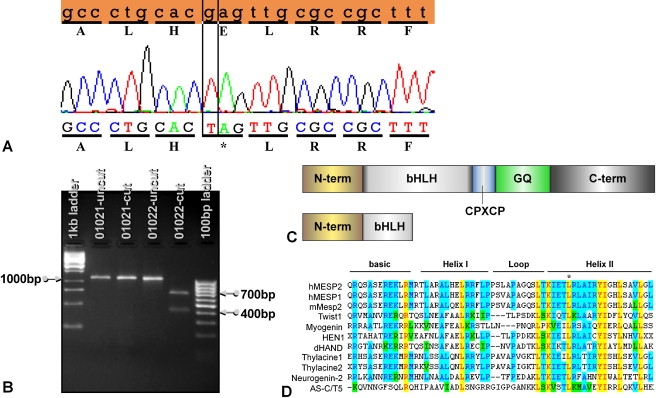
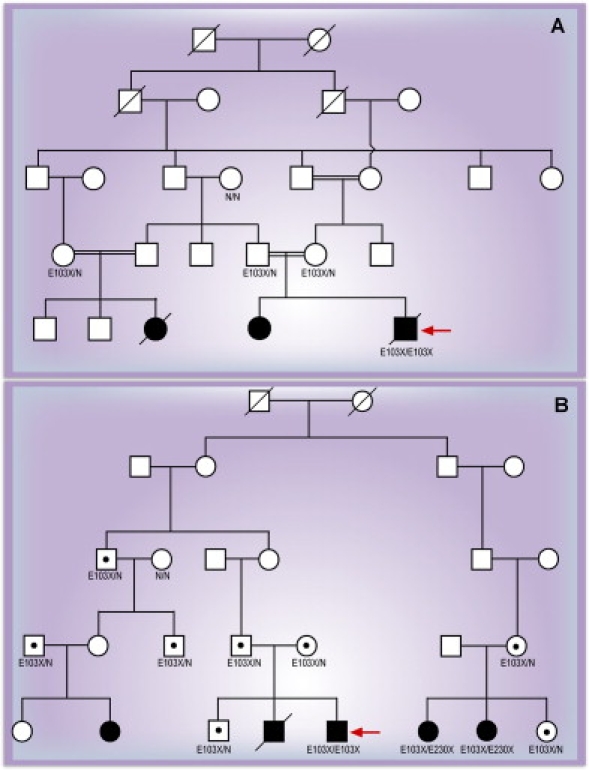
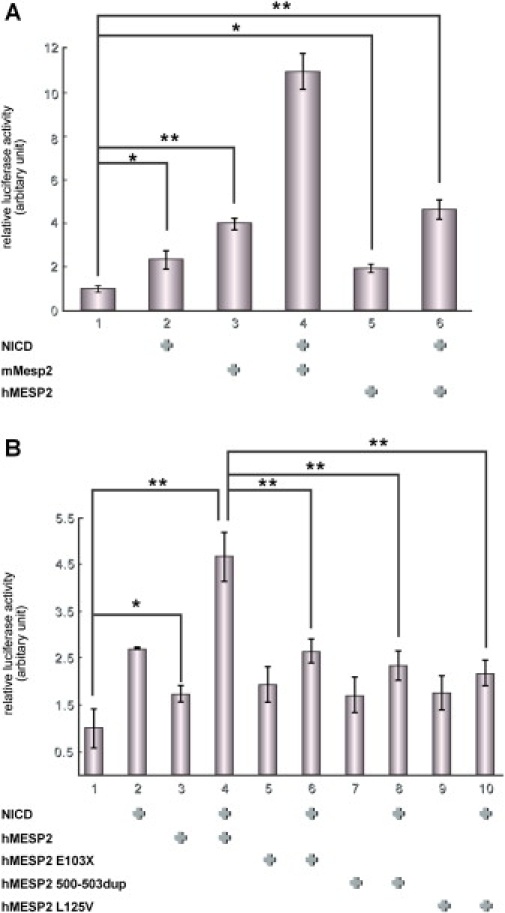
Spondylothoracic dysostosis (STD), also known as Jarcho-Levin syndrome (JLS), is an autosomal-recessive disorder characterized by abnormal vertebral segmentation and defects affecting spine formation, with complete bilateral fusion of the ribs at the costovertebral junction producing a "crab-like" configuration of the thorax. The shortened spine and trunk can severely affect respiratory function during early childhood. The condition is prevalent in the Puerto Rican population, although it is a panethnic disorder. By sequencing a set of candidate genes involved in mouse segmentation, we identified a recessive E103X nonsense mutation in the mesoderm posterior 2 homolog (MESP2) gene in a patient, of Puerto Rican origin and from the Boston area, who had been diagnosed with STD/JLS. We then analyzed 12 Puerto Rican families with STD probands for the MESP2 E103X mutation. Ten patients were homozygous for the E103X mutation, three patients were compound heterozygous for a second nonsense mutation, E230X, or a missense mutation, L125V, which affects a conserved leucine residue within the bHLH region. Thus, all affected probands harbored the E103X mutation. Our findings suggest a founder-effect mutation in the MESP2 gene as a major cause of the classical Puerto Rican form of STD/JLS.
Figures
References
-
- Turnpenny P.D., Alman B., Cornier A.S., Giampietro P.F., Offiah A., Tassy O., Pourquie O., Kusumi K., Dunwoodie S. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev. Dyn. 2007;236:1456–1474. - PubMed
-
- Jarcho S., Levin P. Hereditary malformation of the vertebral bodies. Bull. Johns Hopkins Hosp. 1938;62:216–226.
-
- Cornier A.S., Ramirez N., Arroyo S., Acevedo J., Garcia L., Carlo S., Korf B. Phenotype characterization and natural history of spondylothoracic dysplasia syndrome: a series of 27 new cases. Am. J. Med. Genet. 2004;128:120–126. - PubMed
-
- Lavy N.W., Palmer C.G., Merritt A.D. A syndrome of bizarre vertebral anomalies. J. Pediatr. 1966;69:1121–1125. - PubMed
-
- Moseley J.E., Bonforte R.J. Spondylothoracic dysplasia–a syndrome of congenital anomalies. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1969;106:166–169. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
