

Bioinformatic analysis suggests that the Orbivirus VP6 cistron encodes an overlapping gene
- PMID: 18489030
- PMCID: PMC2373779
- DOI: 10.1186/1743-422X-5-48
Bioinformatic analysis suggests that the Orbivirus VP6 cistron encodes an overlapping gene
Abstract
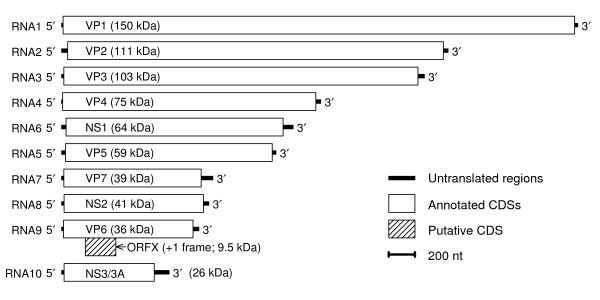
Background: The genus Orbivirus includes several species that infect livestock - including Bluetongue virus (BTV) and African horse sickness virus (AHSV). These viruses have linear dsRNA genomes divided into ten segments, all of which have previously been assumed to be monocistronic.
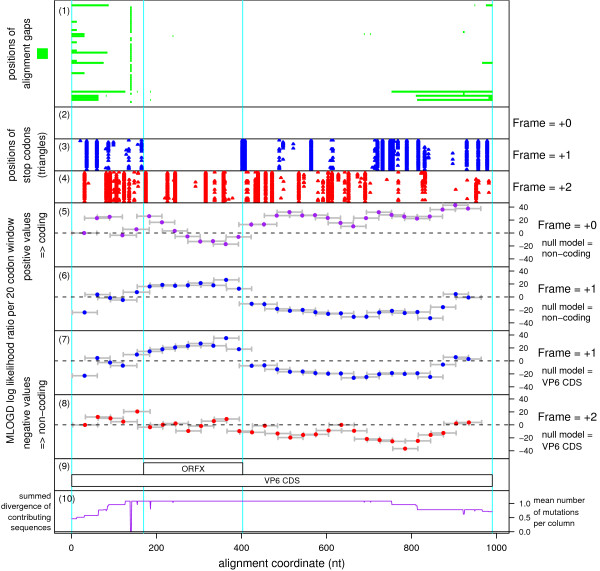
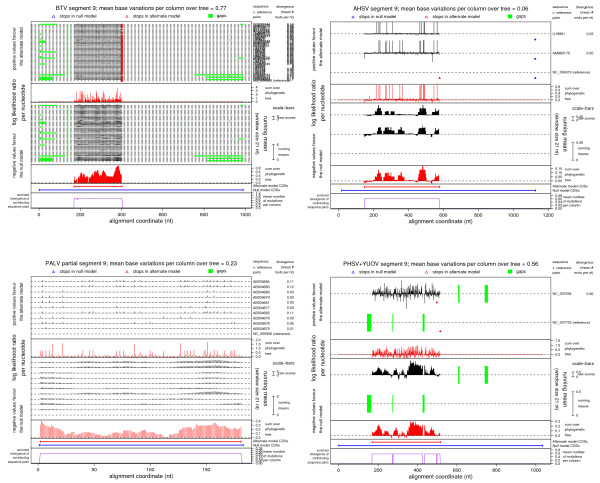
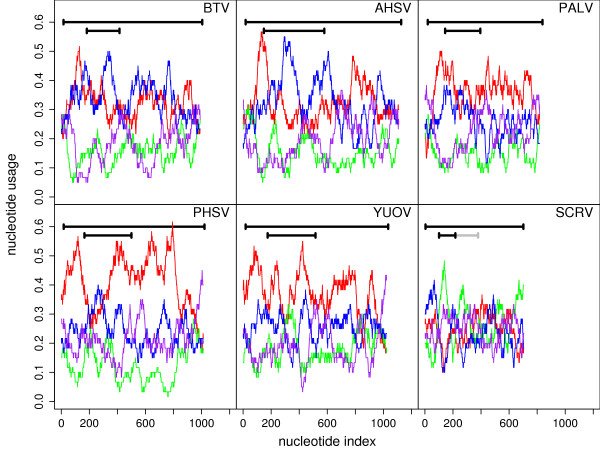
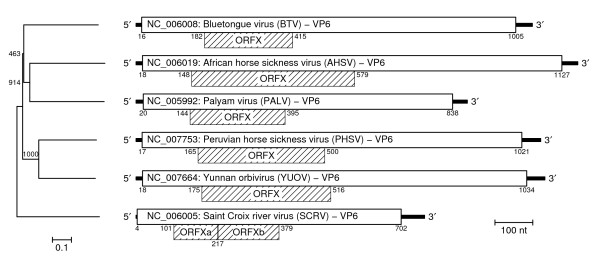
Results: Bioinformatic evidence is presented for a short overlapping coding sequence (CDS) in the Orbivirus genome segment 9, overlapping the VP6 cistron in the +1 reading frame. In BTV, a 77-79 codon AUG-initiated open reading frame (hereafter ORFX) is present in all 48 segment 9 sequences analysed. The pattern of base variations across the 48-sequence alignment indicates that ORFX is subject to functional constraints at the amino acid level (even when the constraints due to coding in the overlapping VP6 reading frame are taken into account; MLOGD software). In fact the translated ORFX shows greater amino acid conservation than the overlapping region of VP6. The ORFX AUG codon has a strong Kozak context in all 48 sequences. Each has only one or two upstream AUG codons, always in the VP6 reading frame, and (with a single exception) always with weak or medium Kozak context. Thus, in BTV, ORFX may be translated via leaky scanning. A long (83-169 codon) ORF is present in a corresponding location and reading frame in all other Orbivirus species analysed except Saint Croix River virus (SCRV; the most divergent). Again, the pattern of base variations across sequence alignments indicates multiple coding in the VP6 and ORFX reading frames.
Conclusion: At approximately 9.5 kDa, the putative ORFX product in BTV is too small to appear on most published protein gels. Nonetheless, a review of past literature reveals a number of possible detections. We hope that presentation of this bioinformatic analysis will stimulate an attempt to experimentally verify the expression and functional role of ORFX, and hence lead to a greater understanding of the molecular biology of these important pathogens.
Figures
References
-
- Attoui H, Mohd Jaafar F, Belhouchet M, Biagini P, Cantaloube JF, de Micco P, de Lamballerie X. Expansion of family Reoviridae to include nine-segmented dsRNA viruses: isolation and characterization of a new virus designated Aedes pseudoscutellaris reovirus assigned to a proposed genus (Dinovernavirus) Virology. 2005;343:212–223. doi: 10.1016/j.virol.2005.08.028. - DOI - PubMed
-
- Landeg F. Bluetongue outbreak in the UK. Vet Rec. 2007;161:534–535. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
