

Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy
- PMID: 18492800
- PMCID: PMC2722888
- DOI: 10.1093/hmg/ddn156
Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy
Abstract
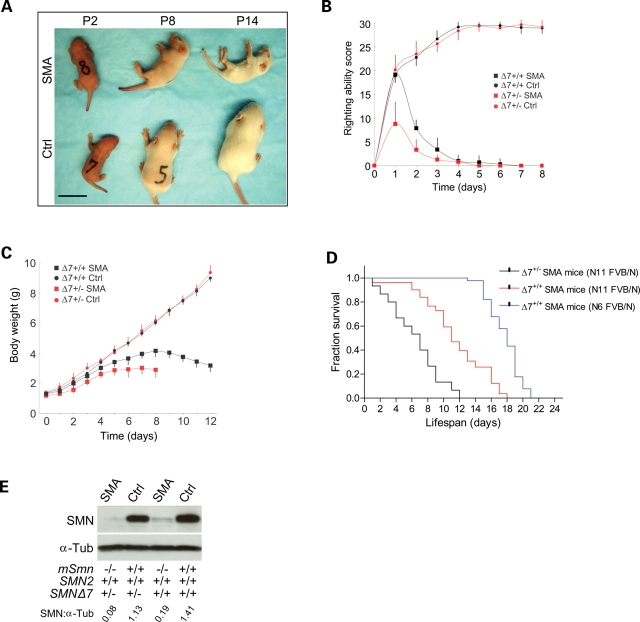
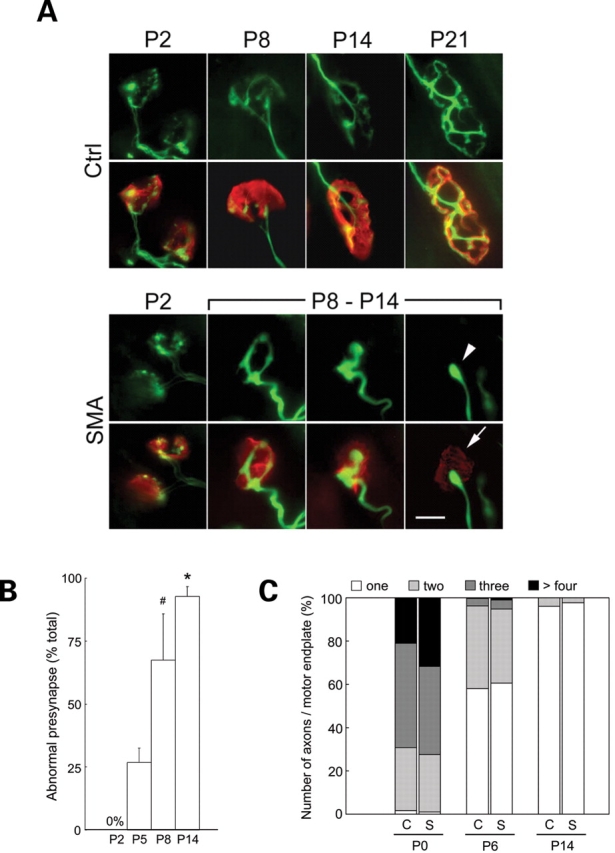
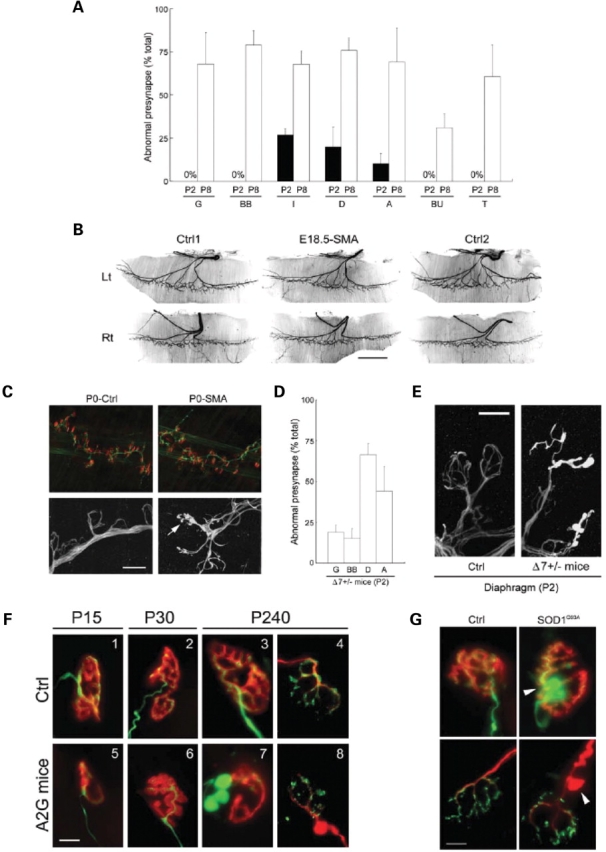
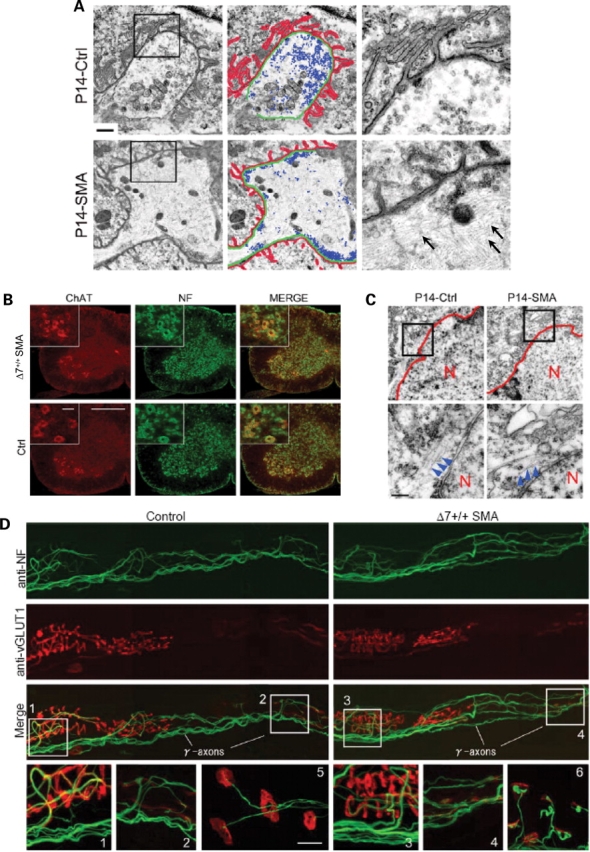
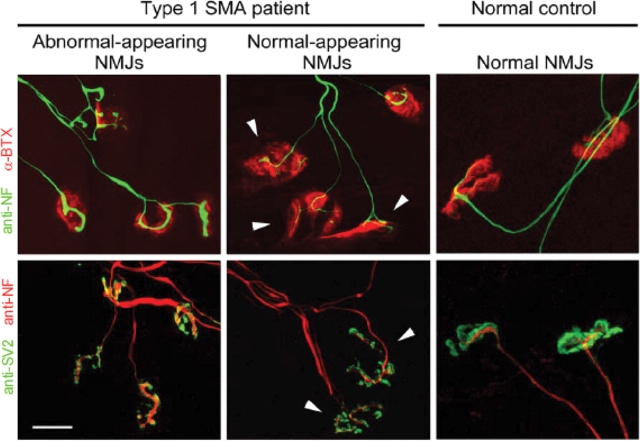
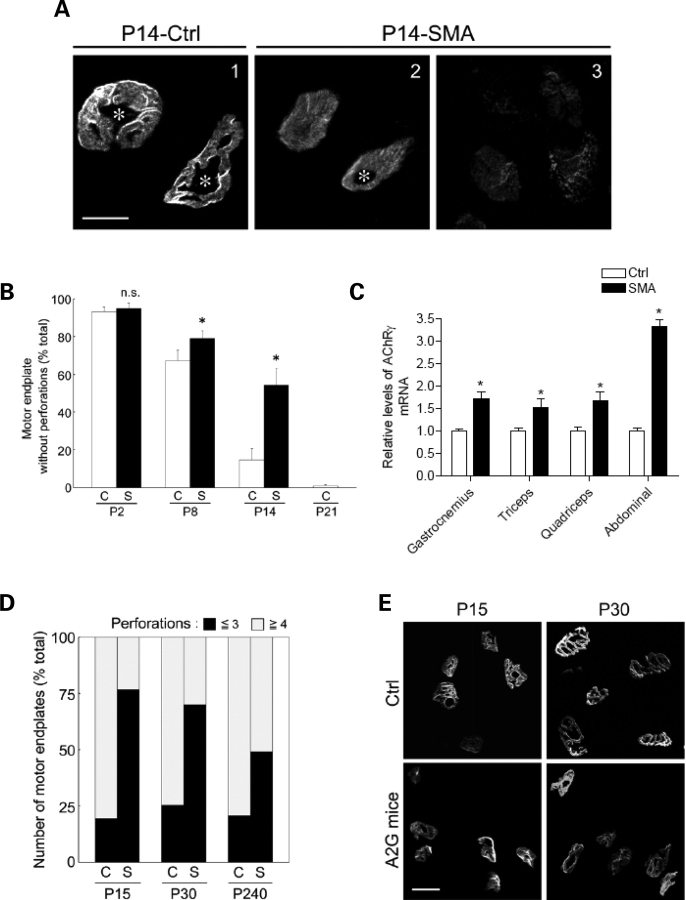
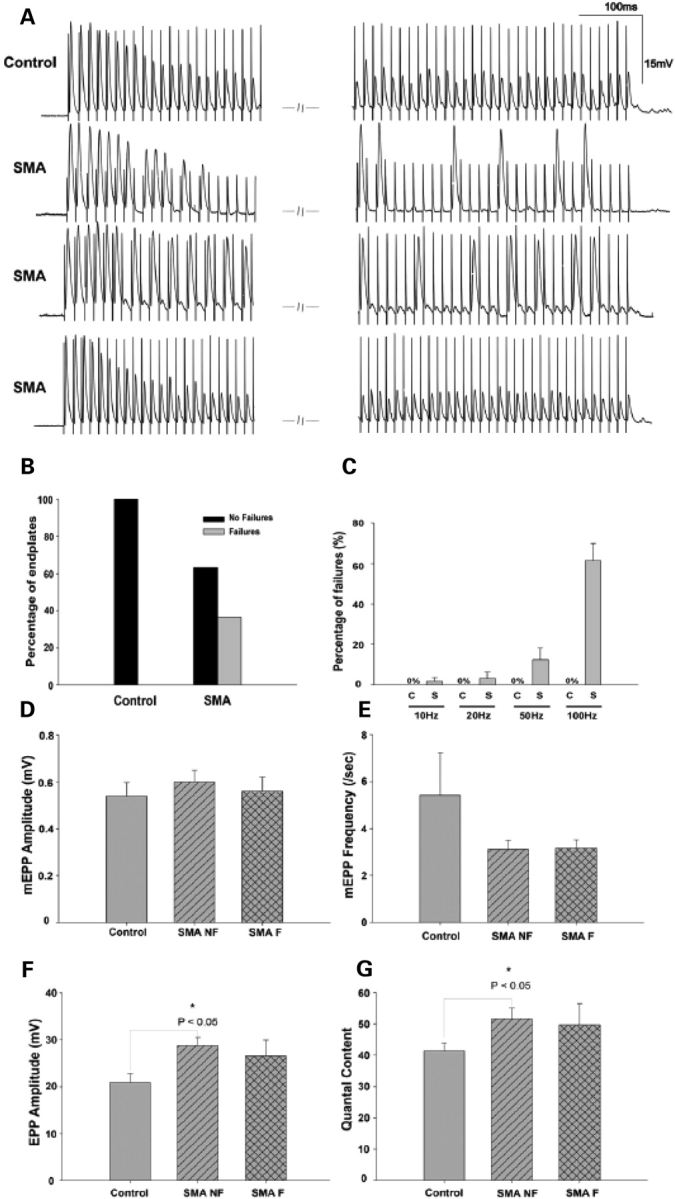
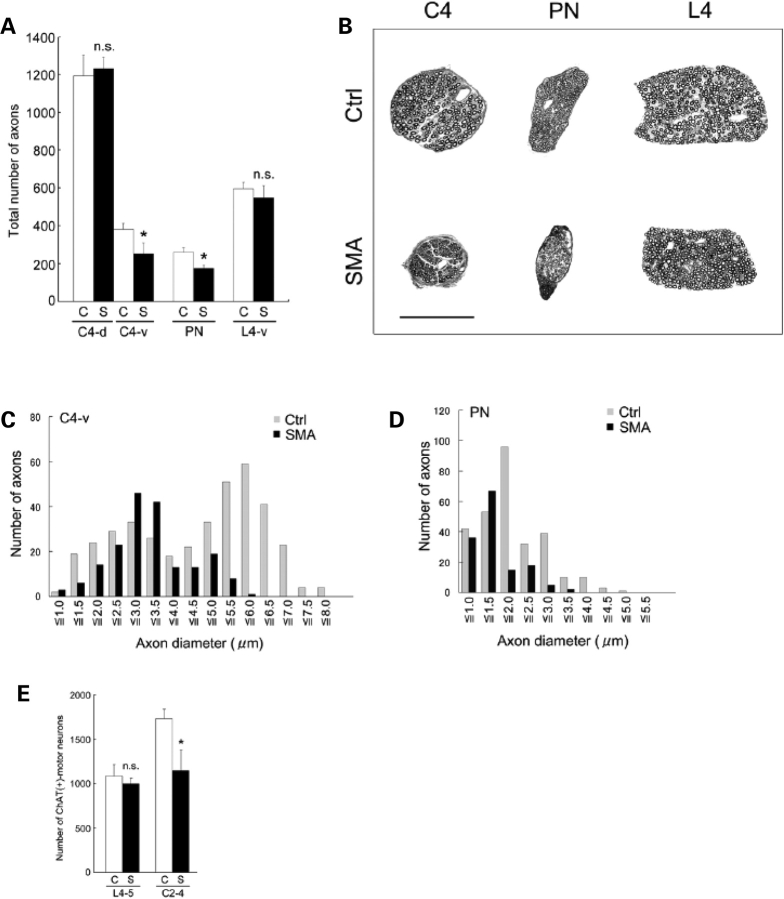
Spinal muscular atrophy (SMA) is a common pediatric neuromuscular disorder caused by insufficient levels of the survival of motor neuron (SMN) protein. Studies involving SMA patients and animal models expressing the human SMN2 gene have yielded relatively little information about the earliest cellular consequences of reduced SMN protein. In this study, we have used severe- and mild-SMN2 expressing mouse models of SMA as well as material from human patients to understand the initial stages of neurodegeneration in the human disease. We show that the earliest structural defects appear distally and involve the neuromuscular synapse. Insufficient SMN protein arrests the post-natal development of the neuromuscular junction (NMJ), impairing the maturation of acetylcholine receptor (AChR) clusters into 'pretzels'. Pre-synaptic defects include poor terminal arborization and intermediate filament aggregates which may serve as a useful biomarker of the disease. These defects are reflected in functional deficits at the NMJ characterized by intermittent neurotransmission failures. We suggest that SMA might best be described as a NMJ synaptopathy and that one promising means of treating it could involve maintaining function at the NMJ.
Figures
Similar articles
-
Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy.Hum Mol Genet. 2008 Apr 1;17(7):949-62. doi: 10.1093/hmg/ddm367. Epub 2007 Dec 8. Hum Mol Genet. 2008. PMID: 18065780
-
Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene.J Neurosci. 2010 Sep 8;30(36):12005-19. doi: 10.1523/JNEUROSCI.2208-10.2010. J Neurosci. 2010. PMID: 20826664 Free PMC article.
-
Motor transmission defects with sex differences in a new mouse model of mild spinal muscular atrophy.EBioMedicine. 2020 May;55:102750. doi: 10.1016/j.ebiom.2020.102750. Epub 2020 Apr 24. EBioMedicine. 2020. PMID: 32339936 Free PMC article.
-
Understanding motoneurone development explains spinal muscular atrophy.Arch Ital Biol. 2007 Nov;145(3-4):325-35. Arch Ital Biol. 2007. PMID: 18075125 Review. No abstract available.
-
Therapeutics development for spinal muscular atrophy.NeuroRx. 2006 Apr;3(2):235-45. doi: 10.1016/j.nurx.2006.01.010. NeuroRx. 2006. PMID: 16554261 Free PMC article. Review.
Cited by
-
Pharmacotherapy for Spinal Muscular Atrophy in Babies and Children: A Review of Approved and Experimental Therapies.Paediatr Drugs. 2022 Nov;24(6):585-602. doi: 10.1007/s40272-022-00529-8. Epub 2022 Aug 27. Paediatr Drugs. 2022. PMID: 36028610 Review.
-
Nusinersen Modulates Proteomics Profiles of Cerebrospinal Fluid in Spinal Muscular Atrophy Type 1 Patients.Int J Mol Sci. 2021 Apr 21;22(9):4329. doi: 10.3390/ijms22094329. Int J Mol Sci. 2021. PMID: 33919289 Free PMC article.
-
Profilin2 regulates actin rod assembly in neuronal cells.Sci Rep. 2021 May 13;11(1):10287. doi: 10.1038/s41598-021-89397-9. Sci Rep. 2021. PMID: 33986363 Free PMC article.
-
Spinal muscular atrophy.Nat Rev Dis Primers. 2022 Aug 4;8(1):52. doi: 10.1038/s41572-022-00380-8. Nat Rev Dis Primers. 2022. PMID: 35927425 Review.
-
Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation.J Clin Invest. 2014 Feb;124(2):785-800. doi: 10.1172/JCI72017. Epub 2014 Jan 27. J Clin Invest. 2014. PMID: 24463453 Free PMC article.
References
-
- Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. - PubMed
-
- Vitali T., Sossi V., Tiziano F., Zappata S., Giuli A., Paravatou-Petsotas M., Neri G., Brahe C. Detection of the survival motor neuron (SMN) genes by FISH: further evidence for a role for SMN2 in the modulation of disease severity in SMA patients. Hum. Mol. Genet. 1999;8:2525–2532. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
