

Splign: algorithms for computing spliced alignments with identification of paralogs
- PMID: 18495041
- PMCID: PMC2440734
- DOI: 10.1186/1745-6150-3-20
Splign: algorithms for computing spliced alignments with identification of paralogs
Abstract
Background: The computation of accurate alignments of cDNA sequences against a genome is at the foundation of modern genome annotation pipelines. Several factors such as presence of paralogs, small exons, non-consensus splice signals, sequencing errors and polymorphic sites pose recognized difficulties to existing spliced alignment algorithms.
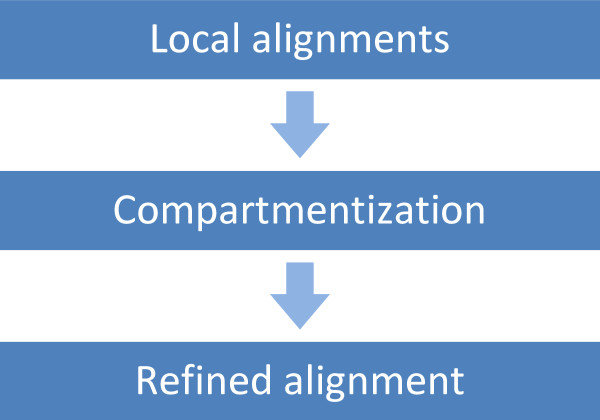
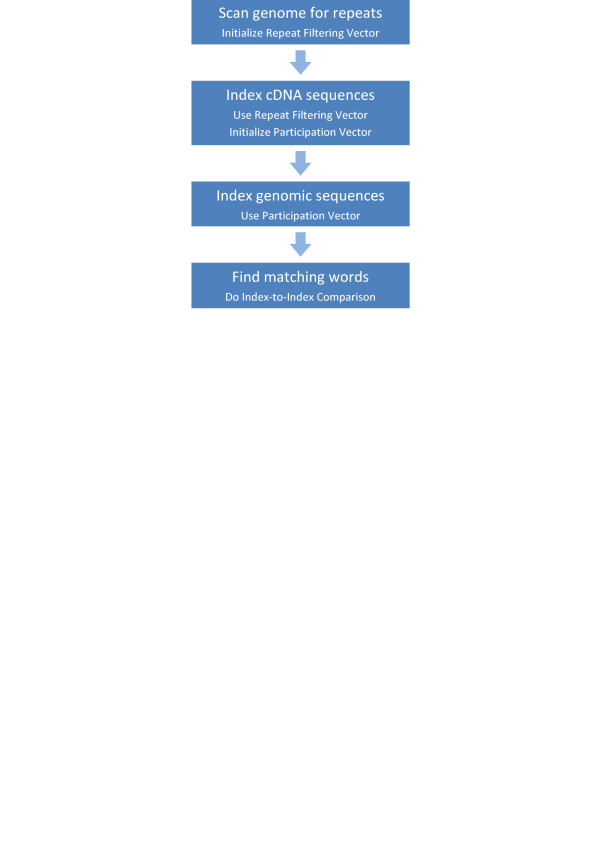
Results: We describe a set of algorithms behind a tool called Splign for computing cDNA-to-Genome alignments. The algorithms include a high-performance preliminary alignment, a compartment identification based on a formally defined model of adjacent duplicated regions, and a refined sequence alignment. In a series of tests, Splign has produced more accurate results than other tools commonly used to compute spliced alignments, in a reasonable amount of time.
Conclusion: Splign's ability to deal with various issues complicating the spliced alignment problem makes it a helpful tool in eukaryotic genome annotation processes and alternative splicing studies. Its performance is enough to align the largest currently available pools of cDNA data such as the human EST set on a moderate-sized computing cluster in a matter of hours. The duplications identification (compartmentization) algorithm can be used independently in other areas such as the study of pseudogenes.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
