

Structure and mechanism of the rebeccamycin sugar 4'-O-methyltransferase RebM
- PMID: 18502766
- PMCID: PMC2504894
- DOI: 10.1074/jbc.M800503200
Structure and mechanism of the rebeccamycin sugar 4'-O-methyltransferase RebM
Abstract
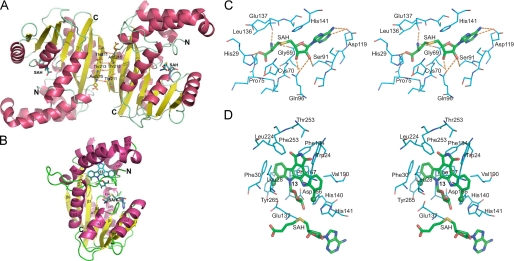
The 2.65-angstroms crystal structure of the rebeccamycin 4'-O-methyltransferase RebM in complex with S-adenosyl-l-homocysteine revealed RebM to adopt a typical S-adenosylmethionine-binding fold of small molecule O-methyltransferases (O-MTases) and display a weak dimerization domain unique to MTases. Using this structure as a basis, the RebM substrate binding model implicated a predominance of nonspecific hydrophobic interactions consistent with the reported ability of RebM to methylate a wide range of indolocarbazole surrogates. This model also illuminated the three putative RebM catalytic residues (His140/141 and Asp166) subsequently found to be highly conserved among sequence-related natural product O-MTases from GC-rich bacteria. Interrogation of these residues via site-directed mutagenesis in RebM demonstrated His140 and Asp166 to be most important for catalysis. This study reveals RebM to be a member of the general acid/base-dependent O-MTases and, as the first crystal structure for a sugar O-MTase, may also present a template toward the future engineering of natural product MTases for combinatorial applications.
Figures



References
-
- Prudhomme, M. (2000) Curr. Med. Chem. 7 1189-1212 - PubMed
-
- Long, B. H., and Balasubramanian, B. N. (2000) Expert Opin. Ther. Pat. 10 635-666
-
- Bailly, C. (2000) Curr. Med. Chem. 7 39-58 - PubMed
-
- Tamaoki, T., Nomoto, H., Takahashi, I., Kato, Y., Morimoto, M., and Tomita, F. (1986) Biochem. Biophys. Res. Commun. 135 397-402 - PubMed
-
- Sanchez, C., Mendez, C., and Salas, J. A. (2006) Nat. Prod. Rep. 23 1007-1045 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
