

Thirteen novel NPHS1 mutations in a large cohort of children with congenital nephrotic syndrome
- PMID: 18503012
- PMCID: PMC2720813
- DOI: 10.1093/ndt/gfn271
Thirteen novel NPHS1 mutations in a large cohort of children with congenital nephrotic syndrome
Abstract
Background: Congenital nephrotic syndrome (CNS) is de- fined as nephrotic syndrome that manifests at birth or within the first 3 months of life. Most patients develop end-stage renal disease (ESRD) within 2 to 3 years of life. CNS of the Finnish-type (CNF) features a rather specific renal histology and is caused by recessive mutations in the NPHS1 gene encoding nephrin, a major structural protein of the glomerular slit-diaphragm. So far, more than 80 different mutations of NPHS1 causing CNF have been published.
Methods: Here, we performed mutation analysis of NPHS1 by exon sequencing in a worldwide cohort of 32 children with CNS from 29 different families.
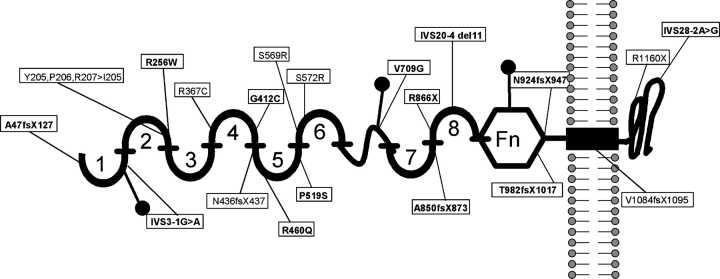
Results: Sixteen of the 29 families (55%) were found to have two disease-causing alleles in NPHS1. Two additional patients had a single heterozygous mutation in NPHS1. Thirteen of a total of 20 different mutations detected were novel (65%). These were five missense mutations, one nonsense mutation, three deletions, one insertion and three splice-site mutations.
Conclusion: Our data expand the spectrum of known NPHS1 mutations by >15% in a worldwide cohort. Surprisingly, two patients with disease-causing mutations showed a relatively mild phenotype, as one patient had a partial remission with steroid treatment and one patient had normal renal function 1 year after the onset of disease. The increased number of known mutations will facilitate future studies into genotype/phenotype correlations.
Figures
References
-
- Norio R. Heredity in the congenital nephrotic syndrome. A genetic study of 57 Finnish families with a review of reported cases. Ann Paediatr Fenn. 1966;12(Suppl 27):1–94. - PubMed
-
- Holmberg C, Trygvasson K, Kestilaa MK, et al. Pediatric Nephrology. 5th edn. Baltimore, MD: Lippincott Williams & Wilkins; 2004. Congenital nephrotic syndrome; pp. 503–516.
-
- Qvist E, Laine J, Ronnholm K, et al. Graft function 5–7 years after renal transplantation in early childhood. Transplantation. 1999;67:1043. - PubMed
-
- Kuusniemi A-M, Merenmies J, Lahdenkari A-T, et al. Glomerular sclerosis in kidneys with congenital nephrotic syndrome (NPHS1) Kidney Int. 2006;70:1423–1431. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
