

Mitochondrial redox signaling by p66Shc is involved in regulating androgenic growth stimulation of human prostate cancer cells
- PMID: 18504439
- PMCID: PMC2776635
- DOI: 10.1038/onc.2008.143
Mitochondrial redox signaling by p66Shc is involved in regulating androgenic growth stimulation of human prostate cancer cells
Abstract
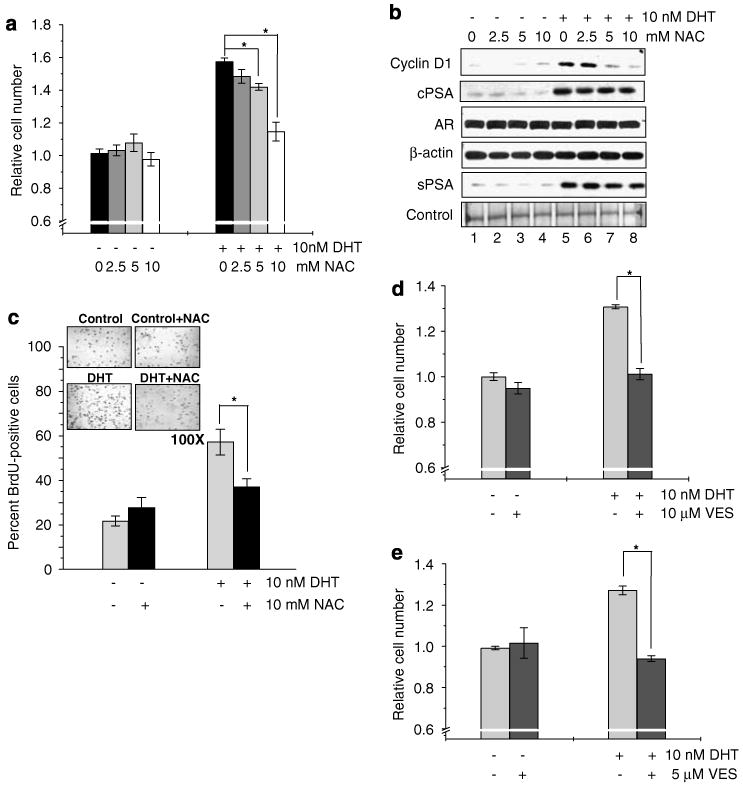
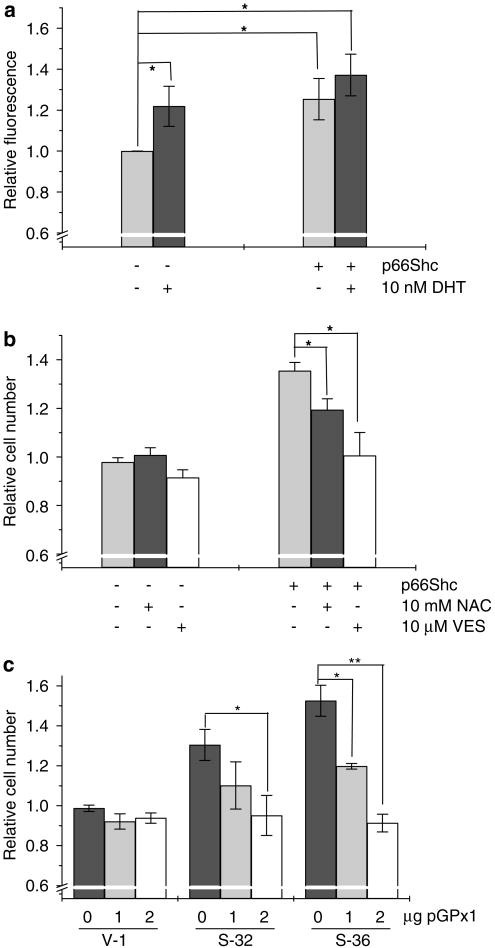
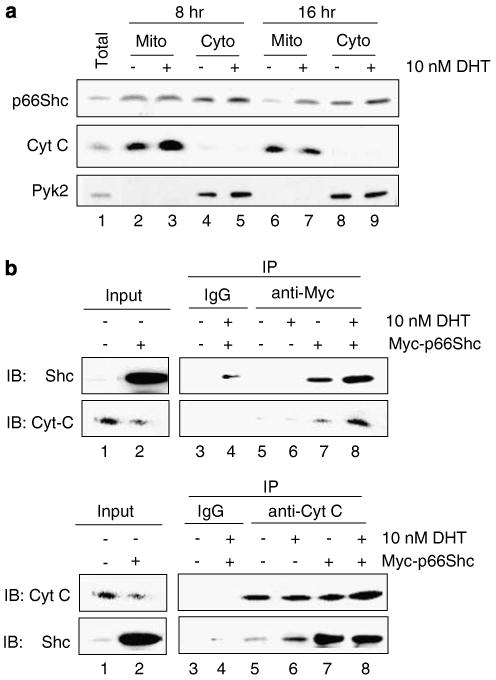
p66Shc is shown to negatively regulate the life span in mice through reactive oxygen species (ROS) production. Recent reports, however, revealed that p66Shc protein level is significantly elevated in several human cancer tissues and growth-stimulated carcinoma cells, suggesting a mitogenic and carcinogenic role for p66Shc. In this communication, we demonstrate for the first time that p66Shc mediates androgenic growth signals in androgen-sensitive human prostate cancer cells through mitochondrial ROS production. Growth stimulation of prostate cancer cells with 5alpha-dihydrotestosterone (DHT) is accompanied by increased p66Shc level and ROS production, which is abolished by antioxidant treatments. However, antioxidant treatments do not affect the transcriptional activity of androgen receptor (AR) as observed by its inability to block DHT-induced prostate-specific antigen expression, an AR-dependent correlate of prostate cancer progression. Elevated expression of p66Shc by cDNA transfection increases the basal cell proliferation and, thus, reduces additional DHT-induced cell proliferation. Furthermore, DHT increases the translocation of p66Shc into mitochondria and its interaction with cytochrome c. Conversely, both redox-negative p66Shc mutant (W134F), which is deficient in cytochrome c interaction, and p66Shc small interfering RNA decrease DHT-induced cell proliferation. These results collectively reveal a novel role for p66Shc-ROS pathway in androgen-induced prostate cancer cell proliferation and, thus, may play a role in early prostate carcinogenesis.
Figures





References
-
- Davol PA, Bagdasaryan R, Elfenbein GJ, Maizel AL, Frackelton AR., Jr Shc proteins are strong, independent prognostic markers for both node-negative and node-positive primary breast cancer. Cancer Res. 2003;63:6772–6783. - PubMed
-
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. - PubMed
-
- Grossman SR, Lyle S, Resnick MB, Sabo E, Lis RT, Rosinha E, et al. p66 Shc Tumor Levels Show a Strong Prognostic Correlation with Disease Outcome in Stage IIA Colon Cancer. Clin Cancer Res. 2007;13:5798–5804. - PubMed
-
- Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809–1818. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
