

Do glial cells control pain?
- PMID: 18504511
- PMCID: PMC2394739
- DOI: 10.1017/S1740925X08000100
Do glial cells control pain?
Abstract
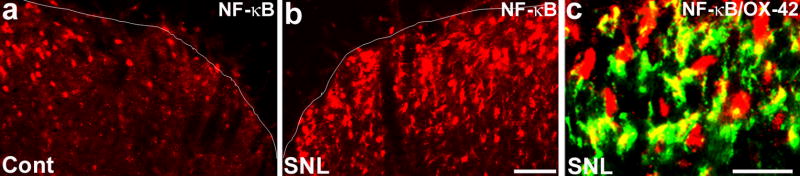
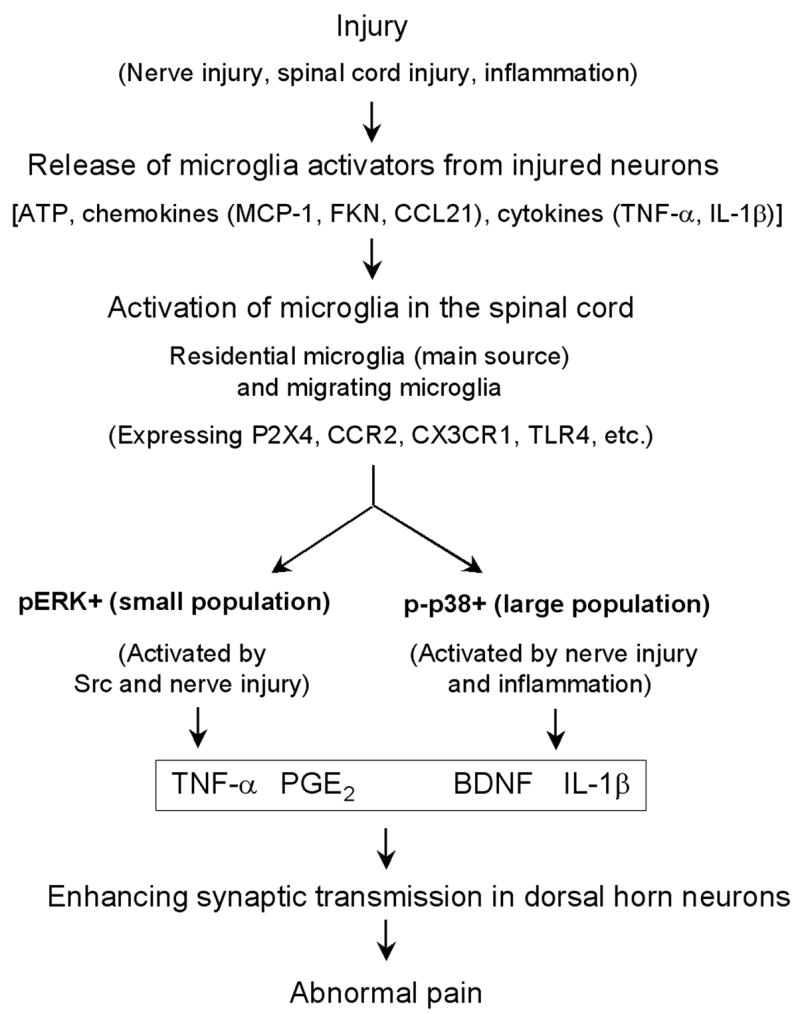
Management of chronic pain is a real challenge, and current treatments that focus on blocking neurotransmission in the pain pathway have resulted in limited success. Activation of glial cells has been widely implicated in neuroinflammation in the CNS, leading to neurodegeneration in conditions such as Alzheimer's disease and multiple sclerosis. The inflammatory mediators released by activated glial cells, such as tumor necrosis factor-a and interleukin-1b not only cause neurodegeneration in these disease conditions, but also cause abnormal pain by acting on spinal cord dorsal horn neurons in injury conditions. Pain can also be potentiated by growth factors such as brain-derived growth factor and basic fibroblast growth factor, which are produced by glia to protect neurons. Thus, glial cells can powerfully control pain when they are activated to produce various pain mediators. We review accumulating evidence that supports an important role for microglial cells in the spinal cord for pain control under injury conditions (e.g. nerve injury). We also discuss possible signaling mechanisms, in particular mitogen-activated protein kinase pathways that are crucial for glial-mediated control of pain.Investigating signaling mechanisms in microglia might lead to more effective management of devastating chronic pain.
Keywords: MAP kinase; chemokines; chronic pain; cytokines; intracellular signaling; microglia; nerve injury.
Figures
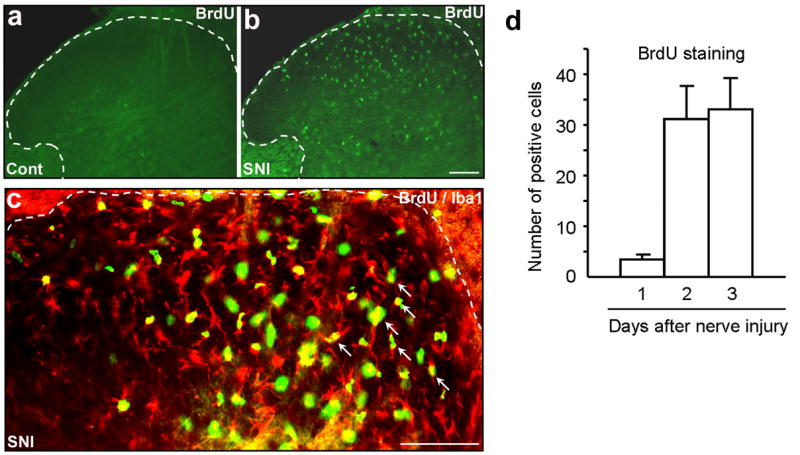
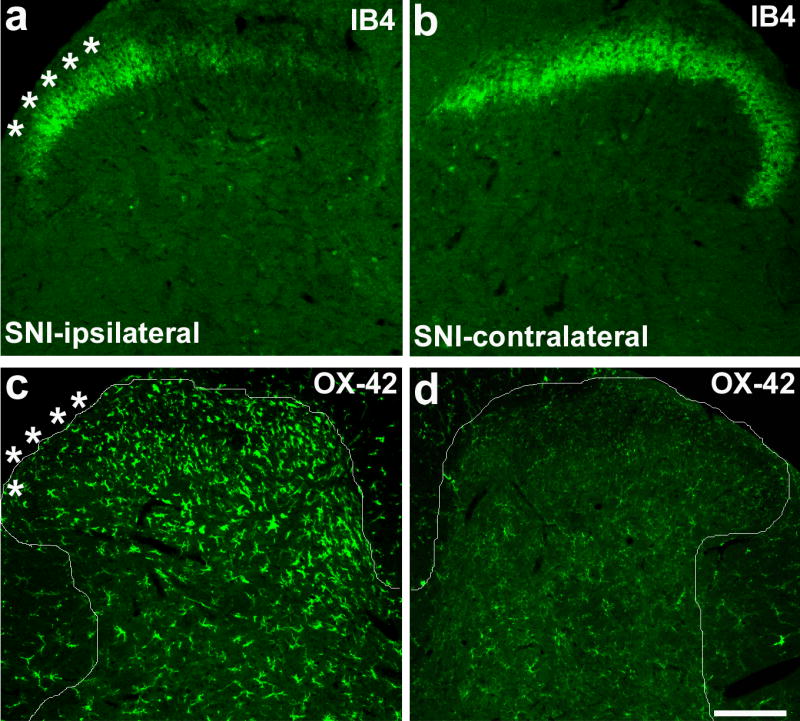
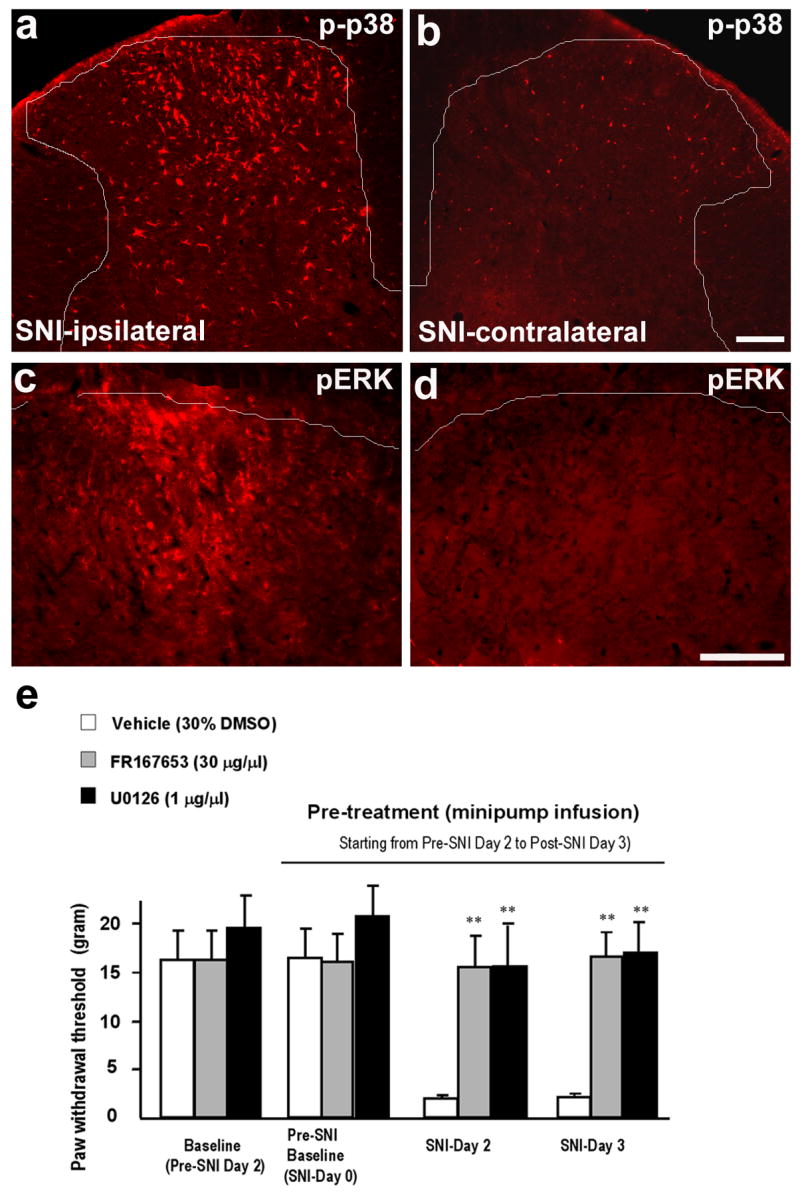
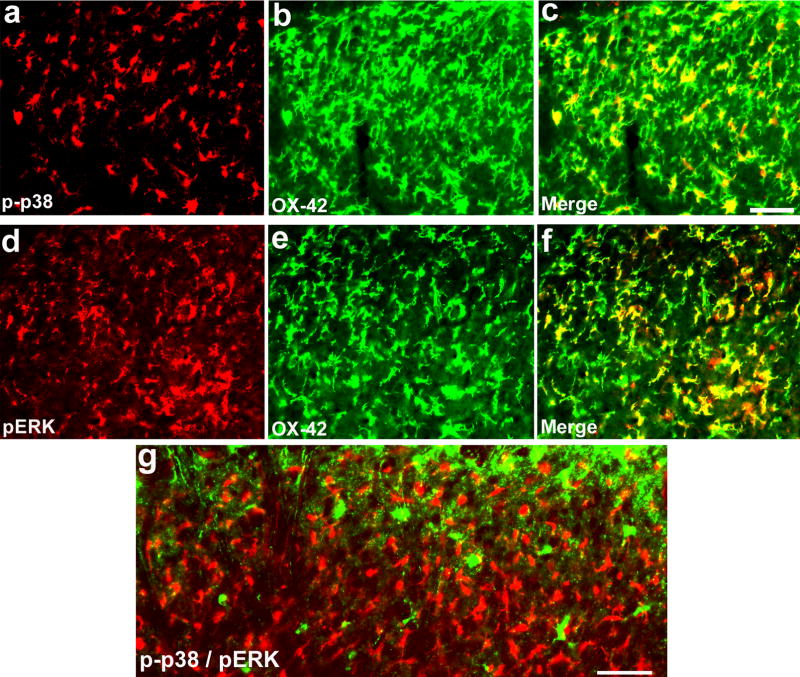
References
-
- Aumeerally N, Allen G, Sawynok J. Glutamate-evoked release of adenosine and regulation of peripheral nociception. Neuroscience. 2004;127(1):1–11. - PubMed
-
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18(5):1633–1641. - PMC - PubMed
-
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical