

Rapid and accurate peptide identification from tandem mass spectra
- PMID: 18505281
- PMCID: PMC2667385
- DOI: 10.1021/pr800127y
Rapid and accurate peptide identification from tandem mass spectra
Abstract
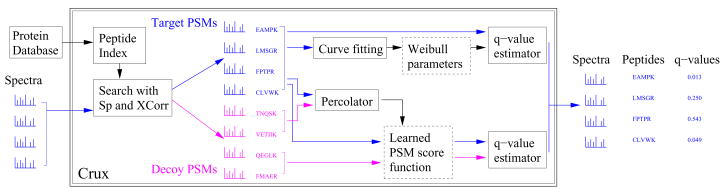
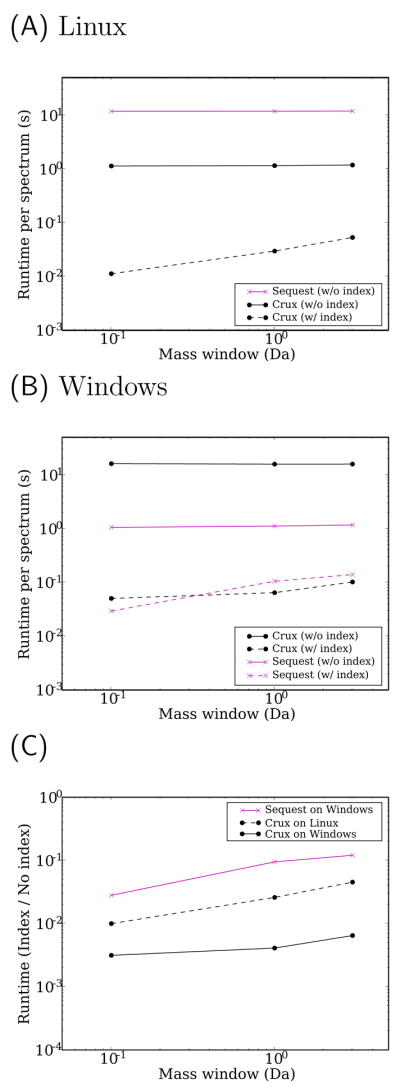
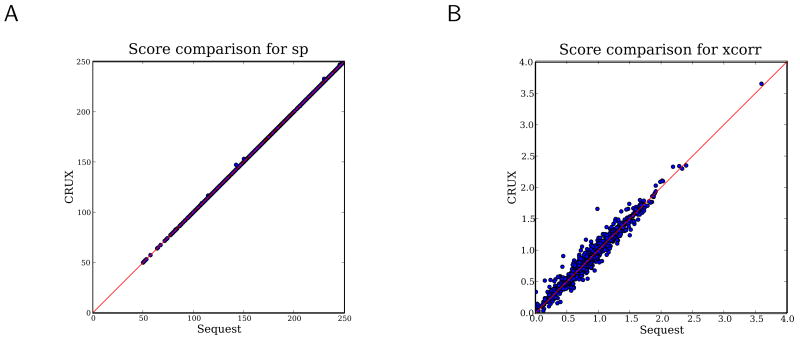
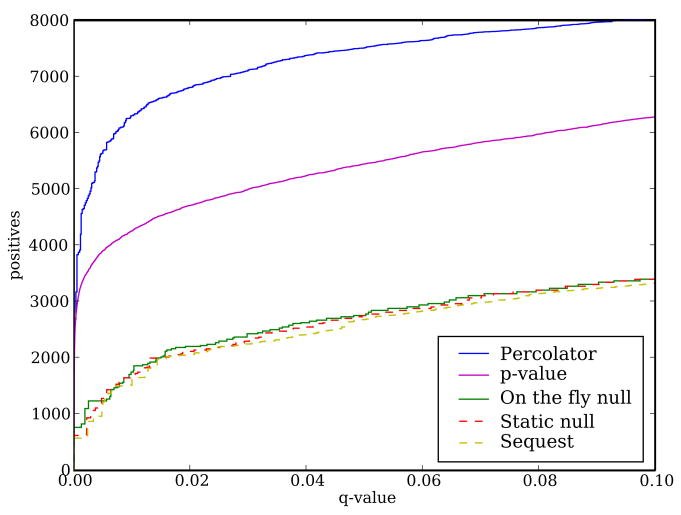
Mass spectrometry, the core technology in the field of proteomics, promises to enable scientists to identify and quantify the entire complement of proteins in a complex biological sample. Currently, the primary bottleneck in this type of experiment is computational. Existing algorithms for interpreting mass spectra are slow and fail to identify a large proportion of the given spectra. We describe a database search program called Crux that reimplements and extends the widely used database search program Sequest. For speed, Crux uses a peptide indexing scheme to rapidly retrieve candidate peptides for a given spectrum. For each peptide in the target database, Crux generates shuffled decoy peptides on the fly, providing a good null model and, hence, accurate false discovery rate estimates. Crux also implements two recently described postprocessing methods: a p value calculation based upon fitting a Weibull distribution to the observed scores, and a semisupervised method that learns to discriminate between target and decoy matches. Both methods significantly improve the overall rate of peptide identification. Crux is implemented in C and is distributed with source code freely to noncommercial users.
Figures
References
-
- Eng JK, McCormack AL, Yates III., JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. - PubMed
-
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. - PubMed
-
- Craig R, Beavis RC. Tandem: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. - PubMed
-
- Tanner S, Shu H, Frank A, Wang LC, Zandi E, et al. InsPecT: Identification of posttranslationally modified peptides from tandem mass spectra. Analytical Chemistry. 2005;77:4626–4639. - PubMed
-
- Bern M, Goldberg D, Cai Y. Lookup peaks: A hybrid de novo sequencing and database search for protein identification by tandem mass spectrometry. Analytical Chemistry. 2007;79:1393–400. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
