

Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death
- PMID: 18505729
- PMCID: PMC2490785
- DOI: 10.1074/jbc.M800214200
Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death
Abstract
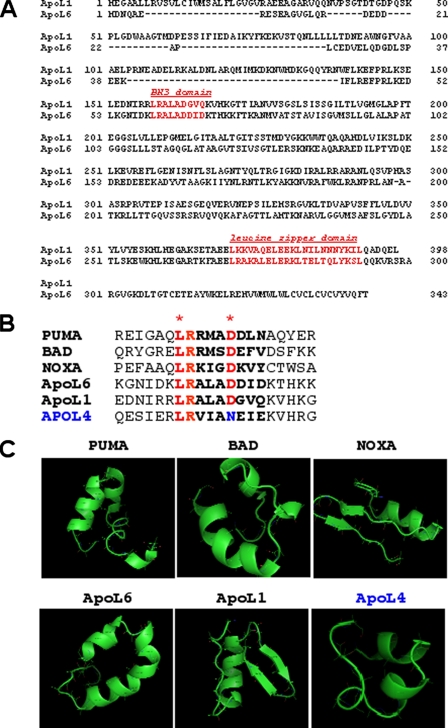
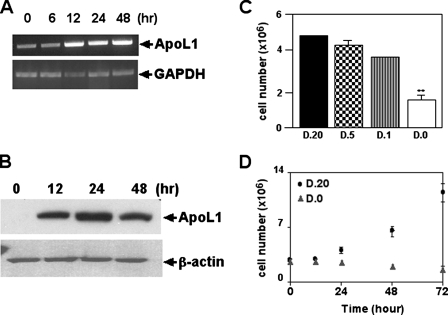
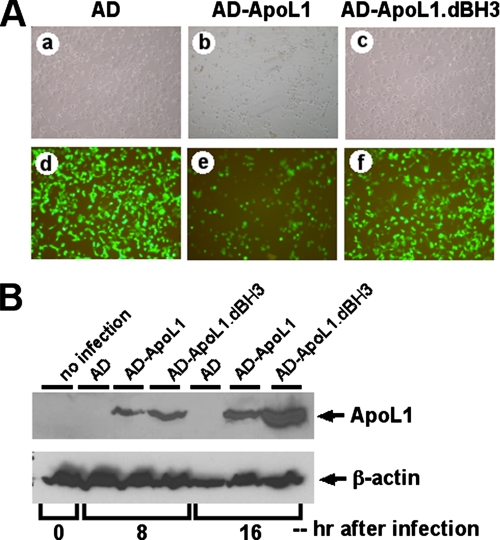
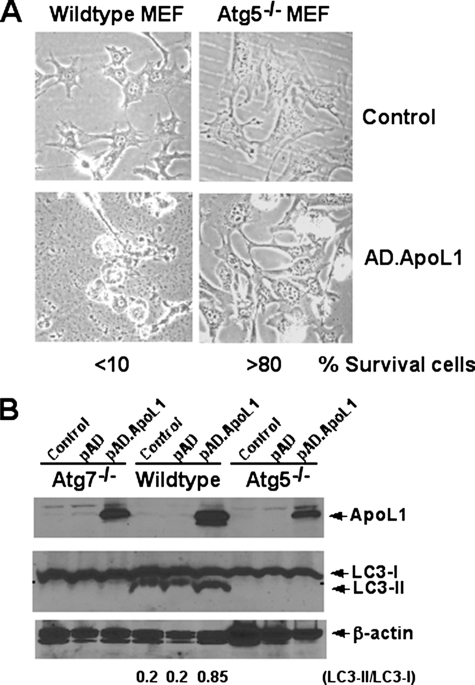
The Bcl-2 family proteins are important regulators of type I programmed cell death apoptosis; however, their role in autophagic cell death (AuCD) or type II programmed cell death is still largely unknown. Here we report the cloning and characterization of a novel Bcl-2 homology domain 3 (BH3)-only protein, apolipoprotein L1 (apoL1), that, when overexpressed and accumulated intracellularly, induces AuCD in cells as characterized by the increasing formation of autophagic vacuoles and activating the translocation of LC3-II from the cytosol to the autophagic vacuoles. Wortmannin and 3-methyladenine, inhibitors of class III phosphatidylinostol 3-kinase and, subsequently, autophagy, blocked apoL1-induced AuCD. In addition, apoL1 failed to induce AuCD in autophagy-deficient ATG5(-/-) and ATG7(-/-) mouse embryonic fibroblast cells, suggesting that apoL1-induced cell death is indeed autophagy-dependent. Furthermore, a BH3 domain deletion construct of apoL1 failed to induce AuCD, demonstrating that apoL1 is a bona fide BH3-only pro-death protein. Moreover, we showed that apoL1 is inducible by p53 in p53-induced cell death and is a lipid-binding protein with high affinity for phosphatidic acid (PA) and cardiolipin (CL). Previously, it has been shown that PA directly interacted with mammalian target of rapamycin and positively regulated the ability of mammalian target of rapamycin to activate downstream effectors. In addition, CL has been shown to activate mitochondria-mediated apoptosis. Sequestering of PA and CL with apoL1 may alter the homeostasis between survival and death leading to AuCD. To our knowledge, this is the first BH3-only protein with lipid binding activity that, when overproduced intracellularly, induces AuCD.
Figures





References
-
- Tsujimoto, Y. (2003) J. Cell. Physiol. 195 158-167 - PubMed
-
- Scorrano, L., and Korsmeyer, S. J. (2003) Biochem. Biophys. Res. Commun. 304 437-444 - PubMed
-
- Huang, D. C., and Strasser, A. (2000) Cell 103 839-842 - PubMed
-
- Chipuk, J. E., Bouchier-Hayes, L., and Green, D. R. (2006) Cell Death Differ. 13 1396-1402 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
