

Whole genome scanning as a cytogenetic tool in hematologic malignancies
- PMID: 18505780
- PMCID: PMC2515145
- DOI: 10.1182/blood-2008-02-130435
Whole genome scanning as a cytogenetic tool in hematologic malignancies
Abstract
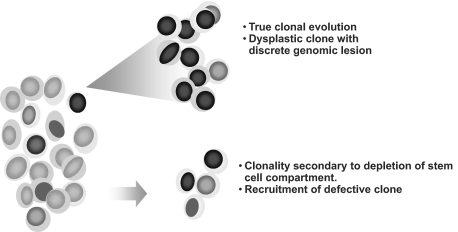
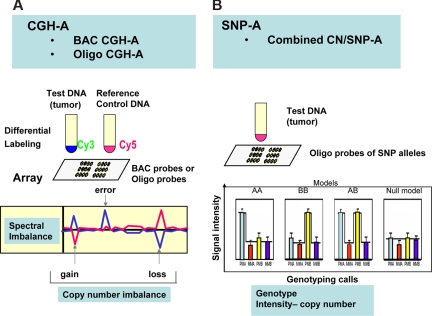
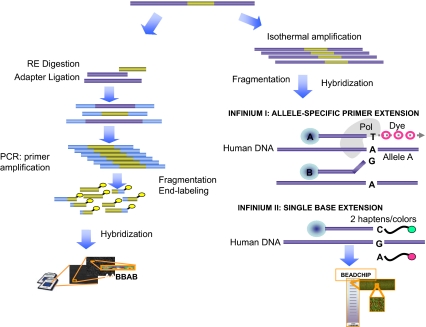
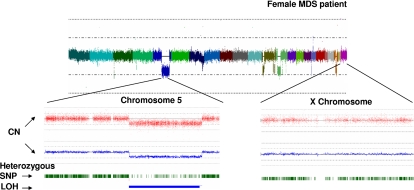
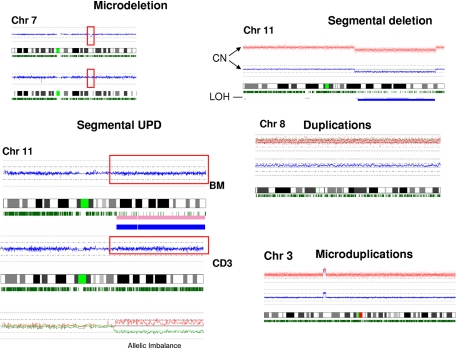
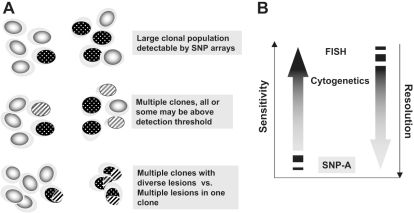
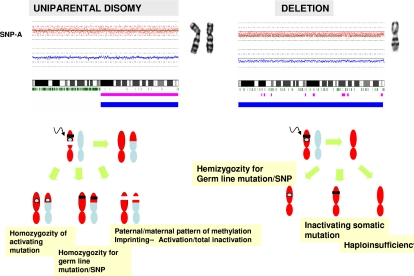
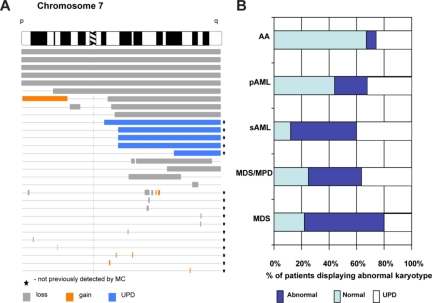
Over the years, methods of cytogenetic analysis evolved and became part of routine laboratory testing, providing valuable diagnostic and prognostic information in hematologic disorders. Karyotypic aberrations contribute to the understanding of the molecular pathogenesis of disease and thereby to rational application of therapeutic modalities. Most of the progress in this field stems from the application of metaphase cytogenetics (MC), but recently, novel molecular technologies have been introduced that complement MC and overcome many of the limitations of traditional cytogenetics, including a need for cell culture. Whole genome scanning using comparative genomic hybridization and single nucleotide polymorphism arrays (CGH-A; SNP-A) can be used for analysis of somatic or clonal unbalanced chromosomal defects. In SNP-A, the combination of copy number detection and genotyping enables diagnosis of copy-neutral loss of heterozygosity, a lesion that cannot be detected using MC but may have important pathogenetic implications. Overall, whole genome scanning arrays, despite the drawback of an inability to detect balanced translocations, allow for discovery of chromosomal defects in a higher proportion of patients with hematologic malignancies. Newly detected chromosomal aberrations, including somatic uniparental disomy, may lead to more precise prognostic schemes in many diseases.
Figures
References
-
- Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1612 patients entered into the MRC AML 10 trial: The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92:2322–2333. - PubMed
-
- Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002;100:4325–4336. - PubMed
-
- Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110:4385–4395. - PubMed
-
- Fenaux P. Chromosome and molecular abnormalities in myelodysplastic syndromes. Int J Hematol. 2001;73:429–437. - PubMed
-
- Sole F, Luno E, Sanzo C, et al. Identification of novel cytogenetic markers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haematologica. 2005;90:1168–1178. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
