

Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria
- PMID: 18507822
- PMCID: PMC2441476
- DOI: 10.1186/gb-2008-9-5-r90
Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria
Abstract
Background: The picocyanobacterial genus Synechococcus occurs over wide oceanic expanses, having colonized most available niches in the photic zone. Large scale distribution patterns of the different Synechococcus clades (based on 16S rRNA gene markers) suggest the occurrence of two major lifestyles ('opportunists'/'specialists'), corresponding to two distinct broad habitats ('coastal'/'open ocean'). Yet, the genetic basis of niche partitioning is still poorly understood in this ecologically important group.
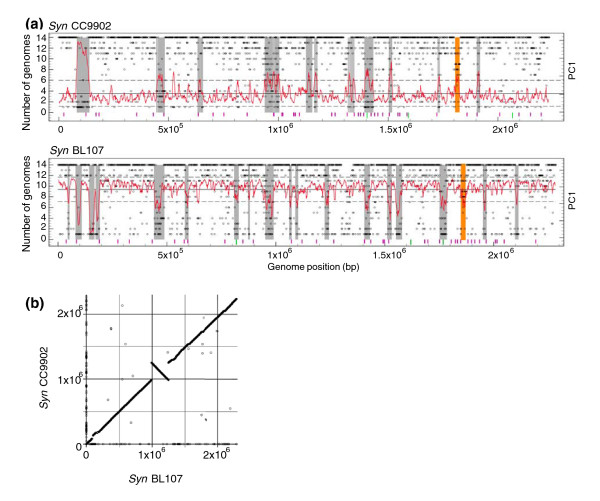
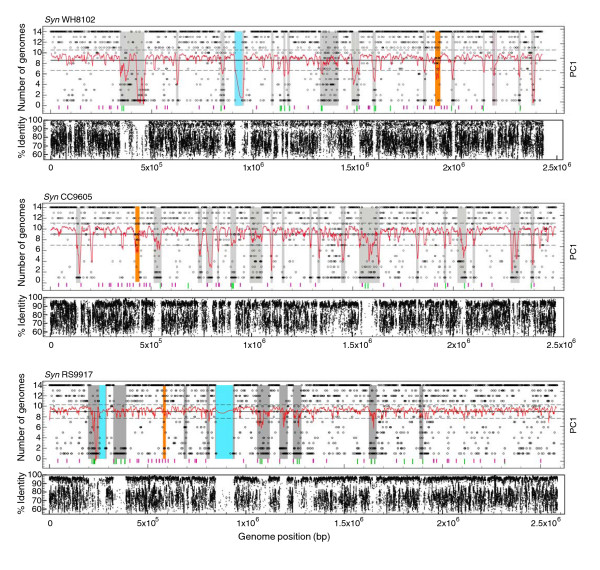
Results: Here, we compare the genomes of 11 marine Synechococcus isolates, representing 10 distinct lineages. Phylogenies inferred from the core genome allowed us to refine the taxonomic relationships between clades by revealing a clear dichotomy within the main subcluster, reminiscent of the two aforementioned lifestyles. Genome size is strongly correlated with the cumulative lengths of hypervariable regions (or 'islands'). One of these, encompassing most genes encoding the light-harvesting phycobilisome rod complexes, is involved in adaptation to changes in light quality and has clearly been transferred between members of different Synechococcus lineages. Furthermore, we observed that two strains (RS9917 and WH5701) that have similar pigmentation and physiology have an unusually high number of genes in common, given their phylogenetic distance.
Conclusion: We propose that while members of a given marine Synechococcus lineage may have the same broad geographical distribution, local niche occupancy is facilitated by lateral gene transfers, a process in which genomic islands play a key role as a repository for transferred genes. Our work also highlights the need for developing picocyanobacterial systematics based on genome-derived parameters combined with ecological and physiological data.
Figures






References
-
- Goericke R, Welschmeyer NA. The marine prochlorophyte Prochlorococcus contributes significantly to phytoplankton biomass and primary production in the Sargasso Sea. Deep-Sea Res. 1993;40:2283–2294. doi: 10.1016/0967-0637(93)90104-B. - DOI
-
- Li WKW. Primary productivity of prochlorophytes, cyanobacteria, and eucaryotic ultraphytoplankton: measurements from flow cytometric sorting. Limnol Oceanogr. 1994;39:169–175.
-
- Moran XAG, Fernandez E, Perez V. Size-fractionated primary production, bacterial production and net community production in subtropical and tropical domains of the oligotrophic NE Atlantic in autumn. Mar Ecol Prog Ser. 2004;274:17–29. doi: 10.3354/meps274017. - DOI
-
- Olson RJ, Zettler ER, Armbrust EV, Chisholm SW. Pigment, size and distribution of Synechococcus in the North Atlantic and Pacific oceans. Limnol Oceanogr. 1990;35:45–58.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
