

Mutations in the TOPORS gene cause 1% of autosomal dominant retinitis pigmentosa
- PMID: 18509552
- PMCID: PMC2391085
Mutations in the TOPORS gene cause 1% of autosomal dominant retinitis pigmentosa
Abstract
Purpose: The purpose of this project was to determine if mutations, including large insertions or deletions, in the recently identified RP31 gene topoisomerase I-binding arginine-serine rich (RS) protein (TOPORS), cause an appreciable fraction of autosomal dominant retinitis pigmentosa (adRP).
Methods: An adRP cohort of 215 families was used to determine the frequency of TOPORS mutations. We looked for mutations in TOPORS by testing 89 probands from the cohort without mutations in other known adRP genes. Mutation detection was performed by fluorescent capillary sequencing and by multiplex ligation probe amplification.
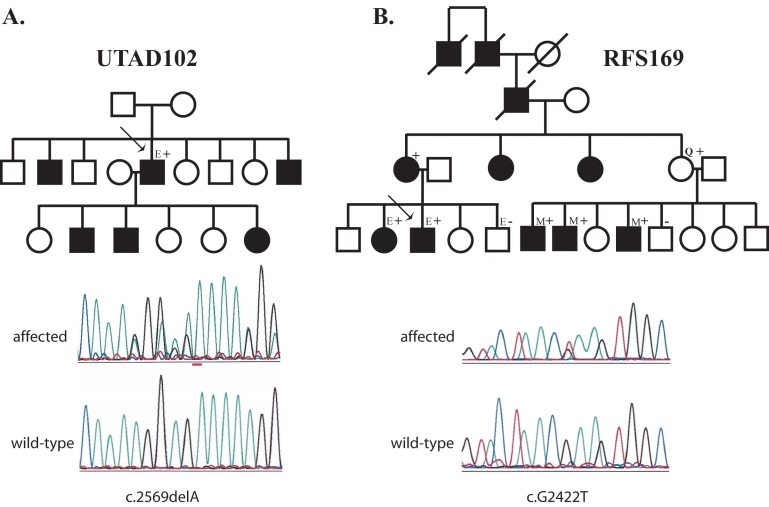
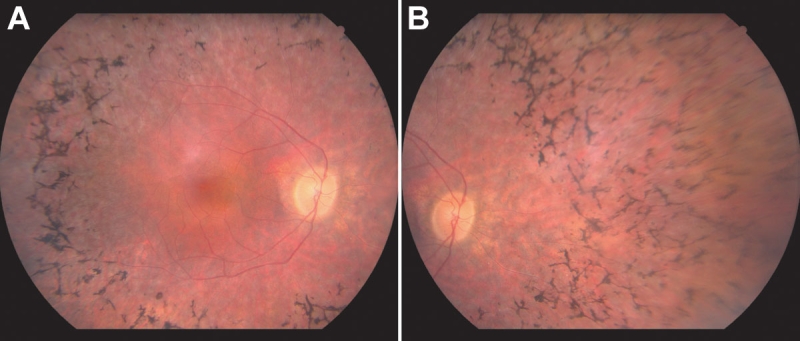
Results: Two different TOPORS mutations, p.Glu808X and p.Arg857GlyfsX9, were each identified in one proband. Patients with these mutations exhibited clinical signs typical of advanced adRP. No large deletions or insertions of TOPORS were identified in our study.
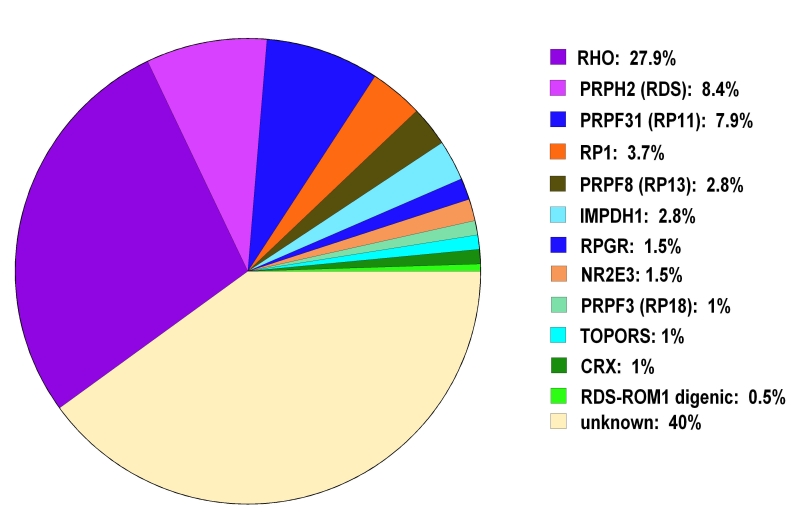
Conclusions: Point mutations and small insertions or deletions in TOPORS cause approximately 1% of adRP. Large deletions or insertions of TOPORS are not an appreciable cause of adRP. Contrary to previous reports, no distinct clinical phenotype was seen in these patients.
Figures
References
-
- Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;(233):1–34. - PubMed
-
- Heckenlively J, Daiger S. 2002 Hereditary retinal and choroidal degenerations, pp 3555–3593 in Emery and Rimoin’s Principles and Practices of Medical Genetics, Vol. 3. Chapter 137. edited by. Rimoin DL, Connor JM, Pyeritz RE, and Korf BR. Churchill Livingstone, Los Angeles.
-
- Papaioannou M, Chakarova CF, Prescott DC, Waseem N, Theis T, Lopez I, Gill B, Koenekoop RK, Bhattacharya SS. A new locus (RP31) for autosomal dominant retinitis pigmentosa maps to chromosome 9p. Hum Genet. 2005;118:501–3. - PubMed
-
- Chakarova CF, Papaioannou MG, Khanna H, Lopez I, Waseem N, Shah A, Theis T, Friedman J, Maubaret C, Bujakowska K, Veraitch B, Abd El-Aziz MM. Prescott de Q, Parapuram SK, Bickmore WA, Munro PM, Gal A, Hamel CP, Marigo V, Ponting CP, Wissinger B, Zrenner E, Matter K, Swaroop A, Koenekoop RK, Bhattacharya SS. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am J Hum Genet. 2007;81:1098–103. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials