

Engineered bacterial outer membrane vesicles with enhanced functionality
- PMID: 18511069
- PMCID: PMC4617544
- DOI: 10.1016/j.jmb.2008.03.076
Engineered bacterial outer membrane vesicles with enhanced functionality
Abstract
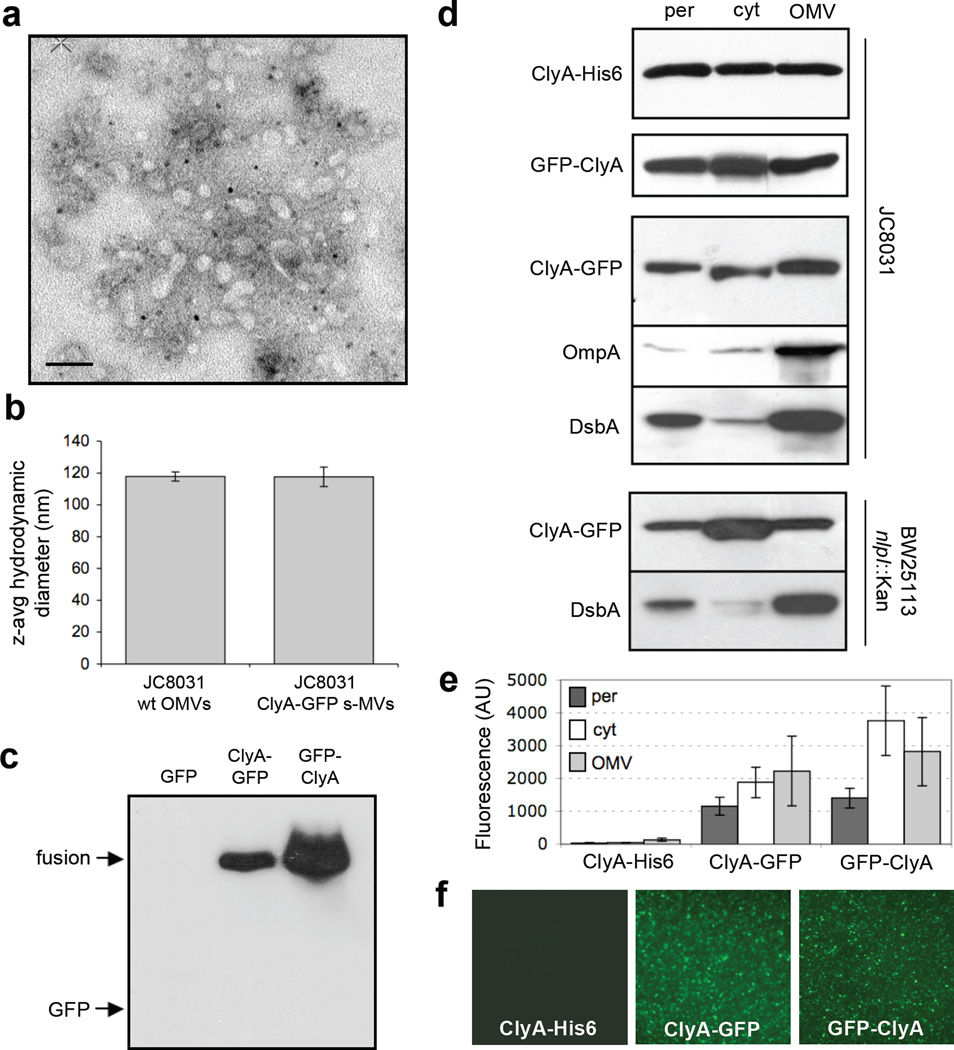
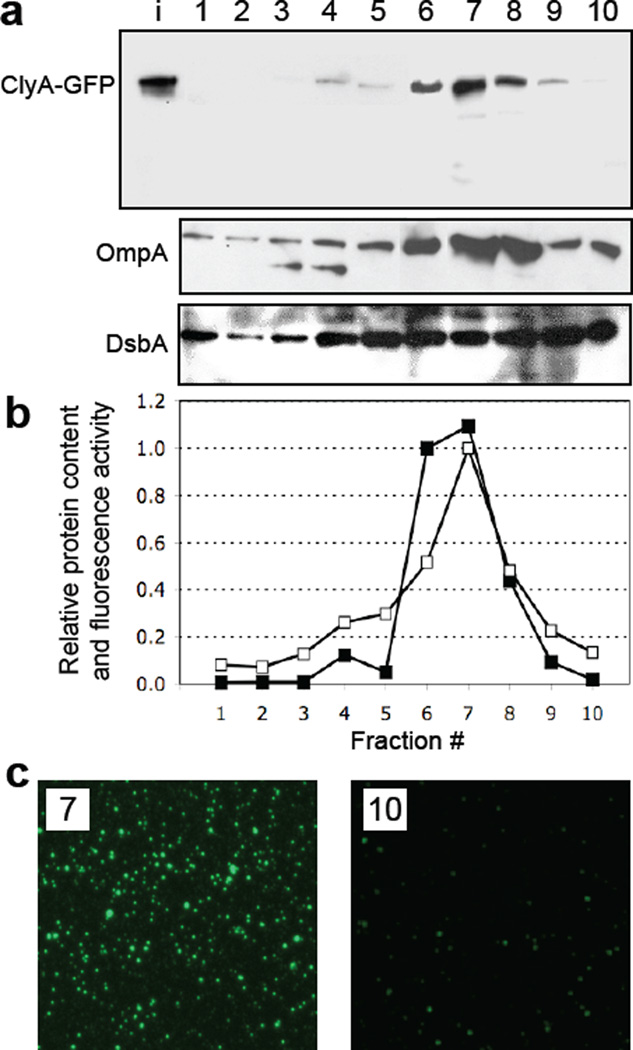
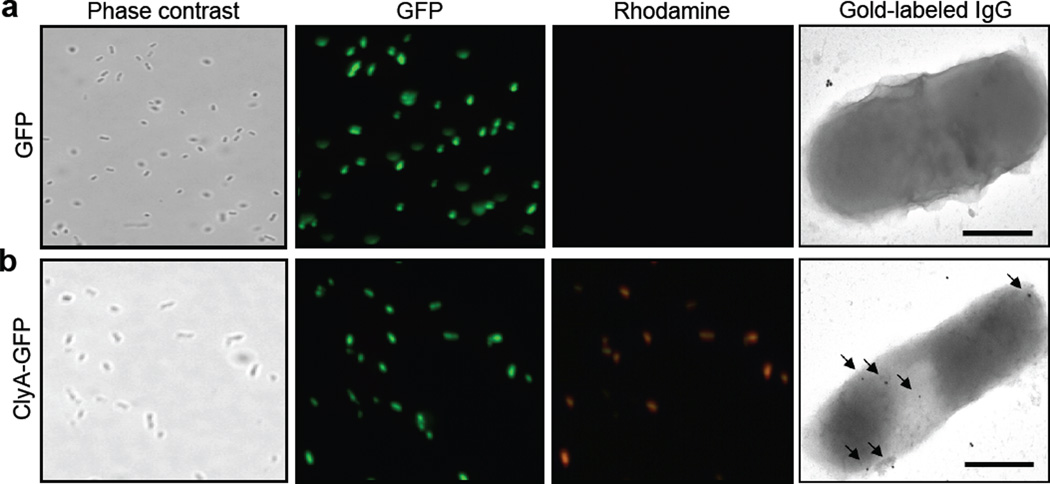
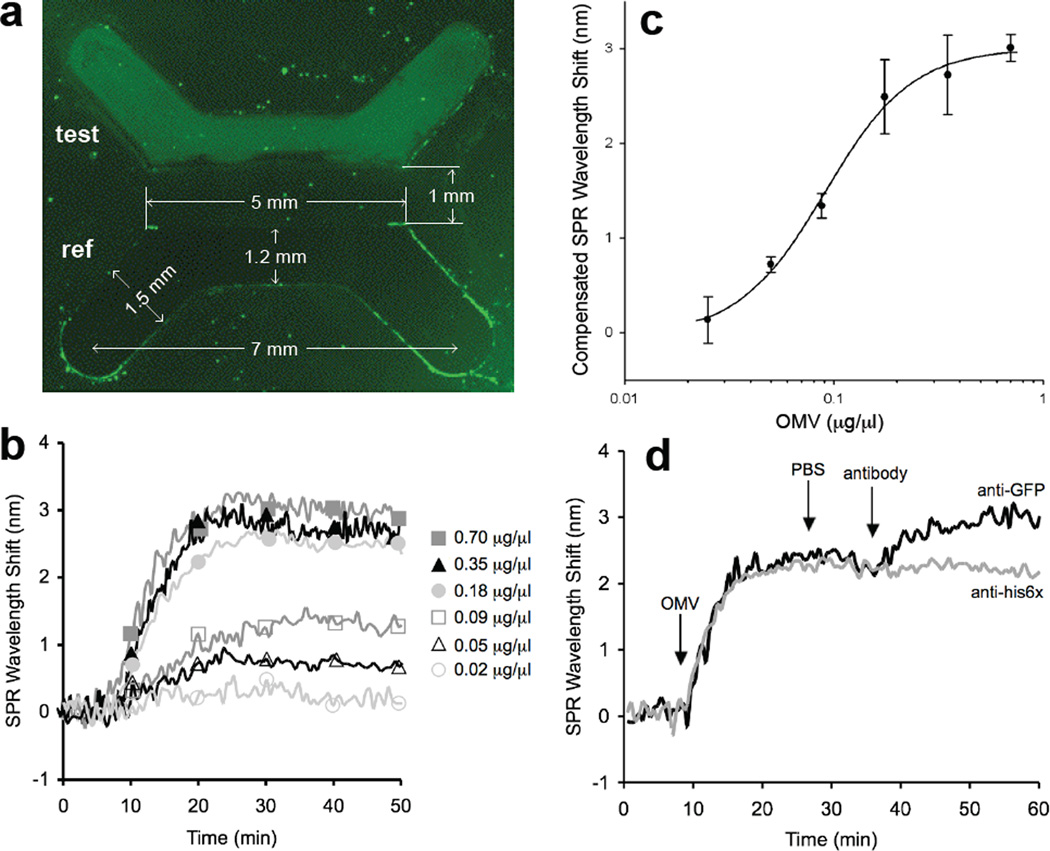
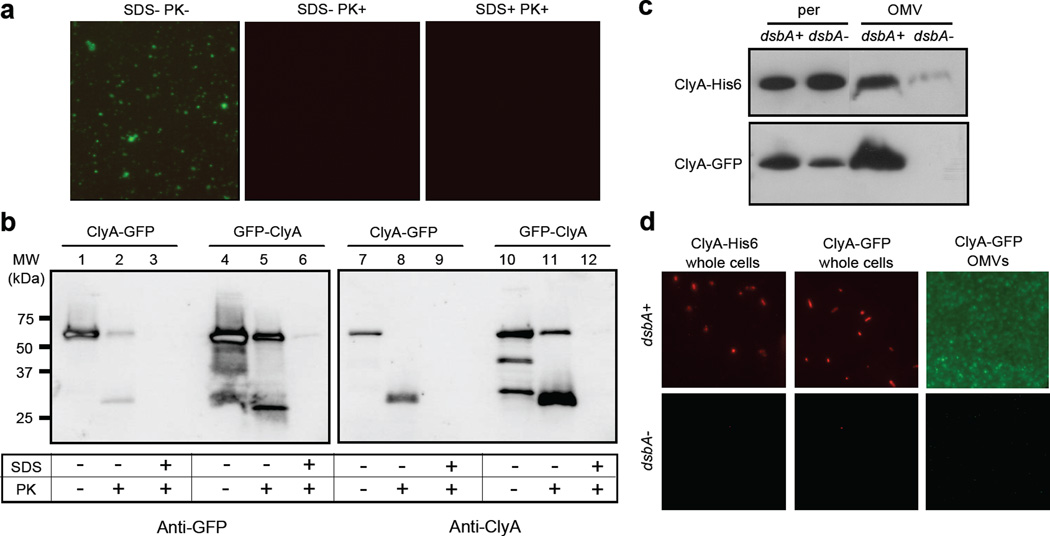
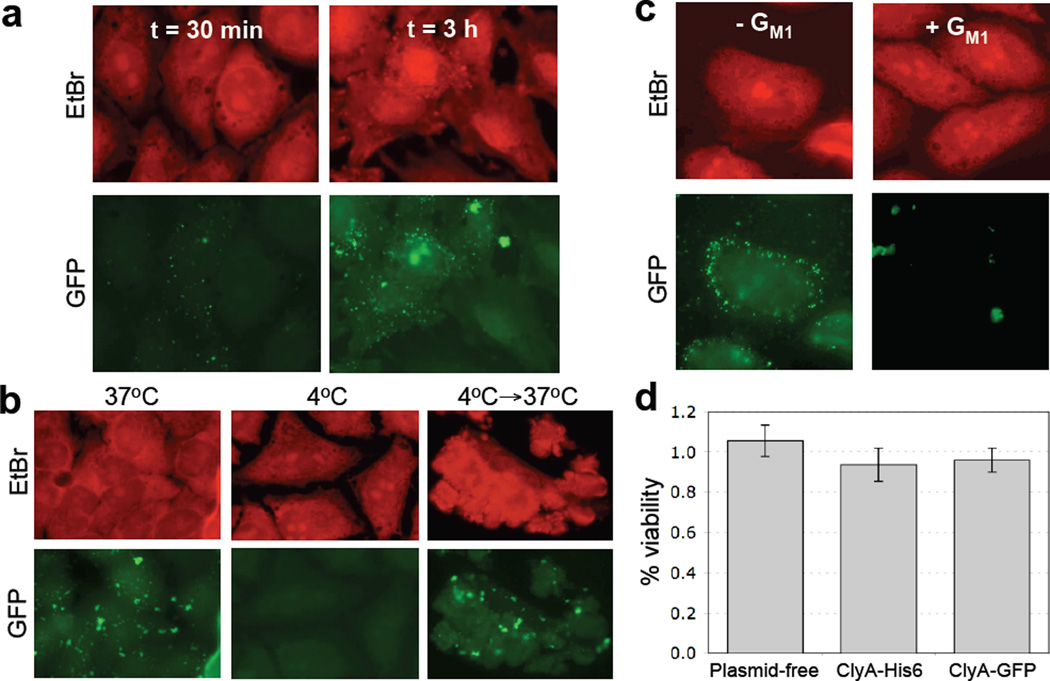
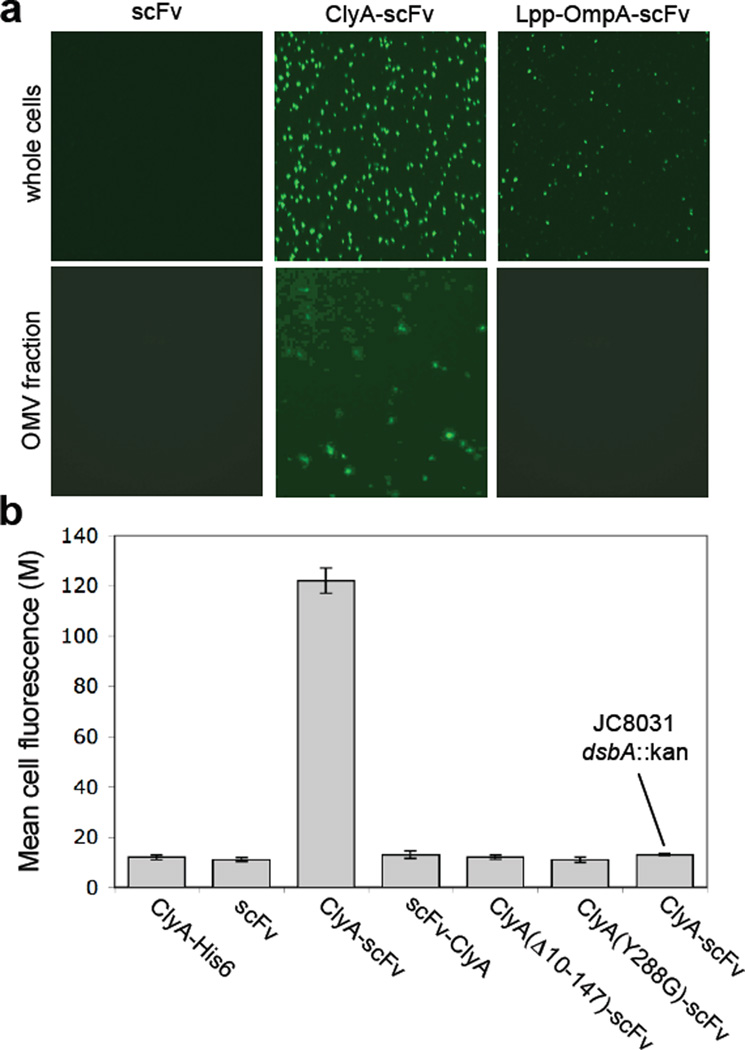
We have engineered bacterial outer membrane vesicles (OMVs) with dramatically enhanced functionality by fusing several heterologous proteins to the vesicle-associated toxin ClyA of Escherichia coli. Similar to native unfused ClyA, chimeric ClyA fusion proteins were found localized in bacterial OMVs and retained activity of the fusion partners, demonstrating for the first time that ClyA can be used to co-localize fully functional heterologous proteins directly in bacterial OMVs. For instance, fusions of ClyA to the enzymes beta-lactamase and organophosphorus hydrolase resulted in synthetic OMVs that were capable of hydrolyzing beta-lactam antibiotics and paraoxon, respectively. Similarly, expression of an anti-digoxin single-chain Fv antibody fragment fused to the C terminus of ClyA resulted in designer "immuno-MVs" that could bind tightly and specifically to the antibody's cognate antigen. Finally, OMVs displaying green fluorescent protein fused to the C terminus of ClyA were highly fluorescent and, as a result of this new functionality, could be easily tracked during vesicle interaction with human epithelial cells. We expect that the relative plasticity exhibited by ClyA as a fusion partner should prove useful for: (i) further mechanistic studies to identify the vesiculation machinery that regulates OMV secretion and to map the intracellular routing of ClyA-containing OMVs during invasion of host cells; and (ii) biotechnology applications such as surface display of proteins and delivery of biologics.
Figures
References
-
- Galan JE, Collmer A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 1999;284:1322–1328. - PubMed
-
- Gentschev I, Dietrich G, Goebel W. The E. coli alpha-hemolysin secretion system and its use in vaccine development. Trends Microbiol. 2002;10:39–45. - PubMed
-
- Henderson IR, Cappello R, Nataro JP. Autotransporter proteins, evolution and redefining protein secretion. Trends Microbiol. 2000;8:529–532. - PubMed
-
- Russel M. Macromolecular assembly and secretion across the bacterial cell envelope: type II protein secretion systems. J Mol Biol. 1998;279:485–499. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
