

Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis
- PMID: 18513679
- PMCID: PMC2427316
- DOI: 10.1016/j.ajhg.2008.05.006
Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis
Erratum in
- Am J Hum Genet. 2008 Aug;83(2):293
Abstract
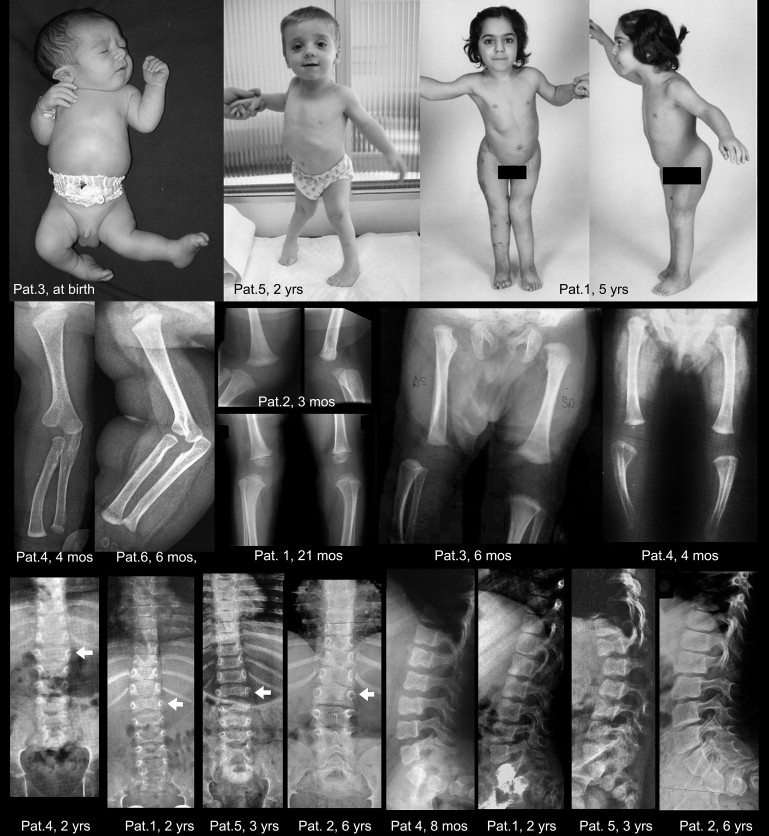
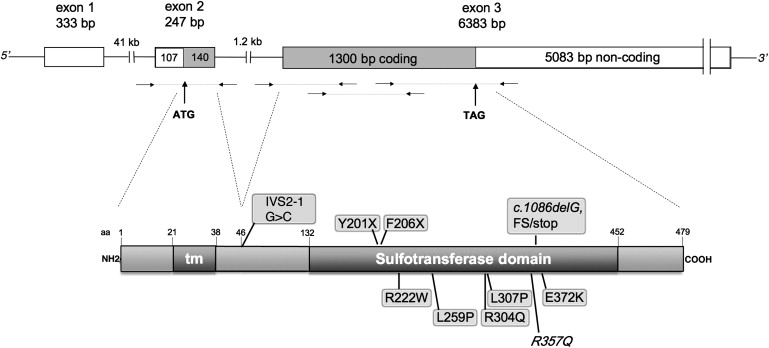
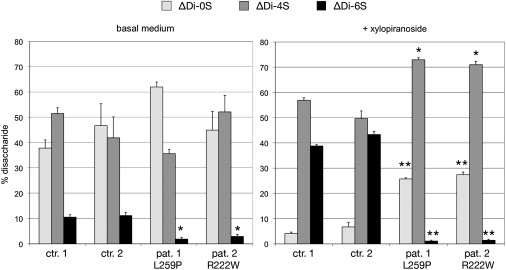
Deficiency of carbohydrate sulfotransferase 3 (CHST3; also known as chondroitin-6-sulfotransferase) has been reported in a single kindred so far and in association with a phenotype of severe chondrodysplasia with progressive spinal involvement. We report eight CHST3 mutations in six unrelated individuals who presented at birth with congenital joint dislocations. These patients had been given a diagnosis of either Larsen syndrome (three individuals) or humero-spinal dysostosis (three individuals), and their clinical features included congenital dislocation of the knees, elbow joint dysplasia with subluxation and limited extension, hip dysplasia or dislocation, clubfoot, short stature, and kyphoscoliosis developing in late childhood. Analysis of chondroitin sulfate proteoglycans in dermal fibroblasts showed markedly decreased 6-O-sulfation but enhanced 4-O-sulfation, confirming functional impairment of CHST3 and distinguishing them from diastrophic dysplasia sulphate transporter (DTDST)-deficient cells. These observations provide a molecular basis for recessive Larsen syndrome and indicate that recessive Larsen syndrome, humero-spinal dysostosis, and spondyloepiphyseal dysplasia Omani type form a phenotypic spectrum.
Figures
References
-
- Raman R., Sasisekharan V., Sasisekharan R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 2005;12:267–277. - PubMed
-
- Thiele H., Sakano M., Kitagawa H., Sugahara K., Rajab A., Hohne W., Ritter H., Leschik G., Nurnberg P., Mundlos S. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. USA. 2004;101:10155–10160. - PMC - PubMed
-
- Rajab A., Kunze J., Mundlos S. Spondyloepiphyseal dysplasia Omani type: A new recessive type of SED with progressive spinal involvement. Am. J. Med. Genet. A. 2004;126:413–419. - PubMed
-
- Larsen L.J., Schottstaedt E.R., Bost F.C. Multiple congenital dislocations associated with characteristic facial abnormality. J. Pediatr. 1950;37:574–581. - PubMed
-
- Latta R.J., Graham C.B., Aase J., Scham S.M., Smith D.W. Larsen's syndrome: A skeletal dysplasia with multiple joint dislocations and unusual facies. J. Pediatr. 1971;78:291–298. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
