

Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome--an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13
- PMID: 18513683
- PMCID: PMC2427271
- DOI: 10.1016/j.ajhg.2008.05.001
Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome--an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13
Abstract
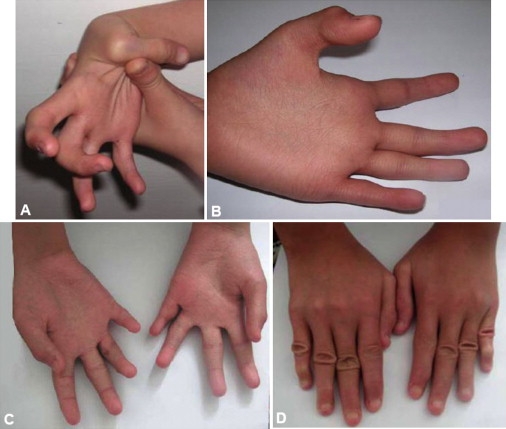
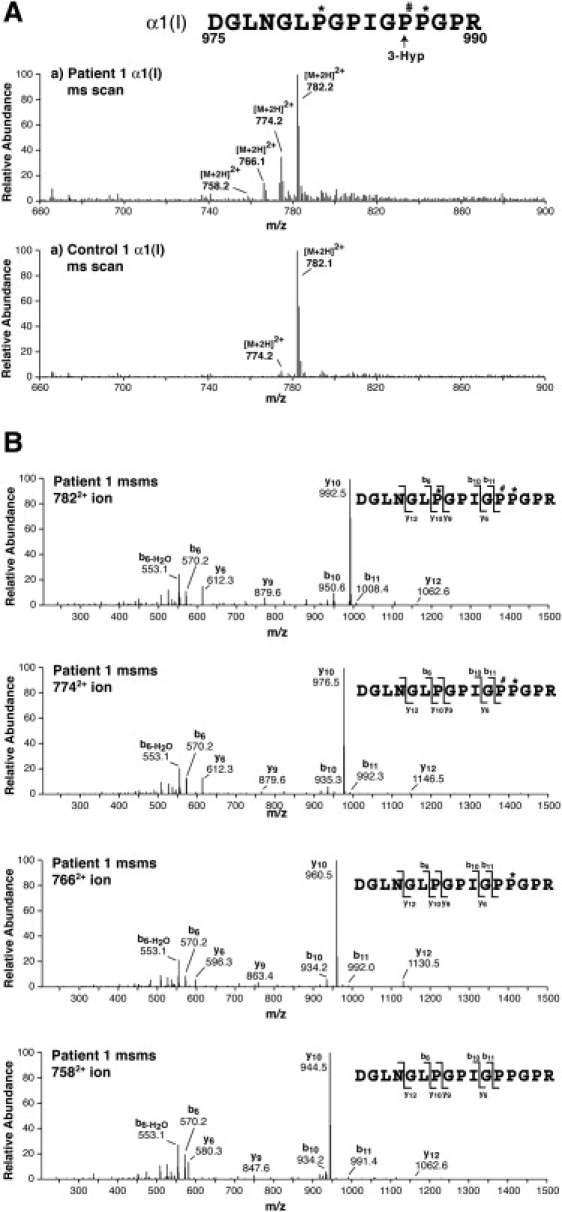
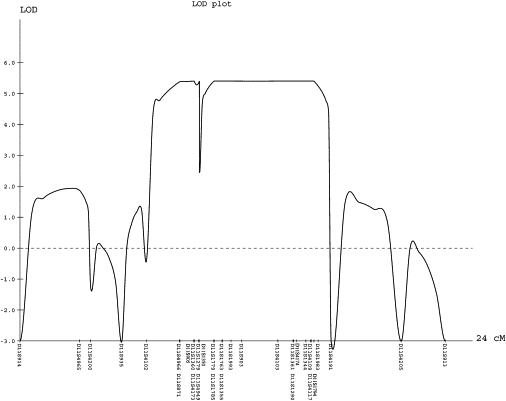
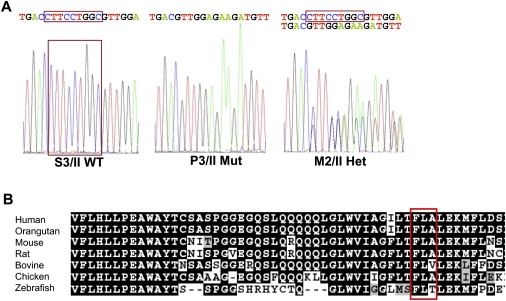
We present clinical, radiological, biochemical, and genetic findings on six patients from two consanguineous families that show EDS-like features and radiological findings of a mild skeletal dysplasia. The EDS-like findings comprise hyperelastic, thin, and bruisable skin, hypermobility of the small joints with a tendency to contractures, protuberant eyes with bluish sclerae, hands with finely wrinkled palms, atrophy of the thenar muscles, and tapering fingers. The skeletal dysplasia comprises platyspondyly with moderate short stature, osteopenia, and widened metaphyses. Patients have an increased ratio of total urinary pyridinolines, lysyl pyridinoline/hydroxylysyl pyridinoline (LP/HP), of approximately 1 as opposed to approximately 6 in EDS VI or approximately 0.2 in controls. Lysyl and prolyl residues of collagens were underhydroxylated despite normal lysyl hydroxylase and prolyl 4-hydroxylase activities; underhydroxylation was a generalized process as shown by mass spectrometry of the alpha1(I)- and alpha2(I)-chain-derived peptides of collagen type I and involved at least collagen types I and II. A genome-wide SNP scan and sequence analyses identified in all patients a homozygous c.483_491 del9 SLC39A13 mutation that encodes for a membrane-bound zinc transporter SLC39A13. We hypothesize that an increased Zn(2+) content inside the endoplasmic reticulum competes with Fe(2+), a cofactor that is necessary for hydroxylation of lysyl and prolyl residues, and thus explains the biochemical findings. These data suggest an entity that we have designated "spondylocheiro dysplastic form of EDS (SCD-EDS)" to indicate a generalized skeletal dysplasia involving mainly the spine (spondylo) and striking clinical abnormalities of the hands (cheiro) in addition to the EDS-like features.
Figures



References
-
- Beighton P., De Paepe A., Steinmann B., Tsipouras P., Wenstrup R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998;77:31–37. - PubMed
-
- Steinmann B., Royce P.M., Superti-Furga A. The Ehlers-Danlos syndrome. In: Royce P.M., Steinmann B., editors. Connective Tissue and its Heritable Disorders. Second Edition. Wiley-Liss; New York: 2002. pp. 431–523.
-
- Kraenzlin, M.E., Kraenzlin, C.A., Meier, C., Giunta, C., and Steinmann, B. Evaluation of an automated HPLC system for measurement of urinary collagen crosslinks: Effect of age, menopause and metabolic bone diseases. Clin. Chem., in press. - PubMed
-
- Giunta C., Randolph A., Al-Gazali L., Brunner H.G., Kraenzlin M.E., Steinmann B. The Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VIA) Am. J. Med. Genet. 2005;133:158–164. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
