

Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1
- PMID: 18515365
- PMCID: PMC2533786
- DOI: 10.1074/jbc.M707692200
Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1
Abstract
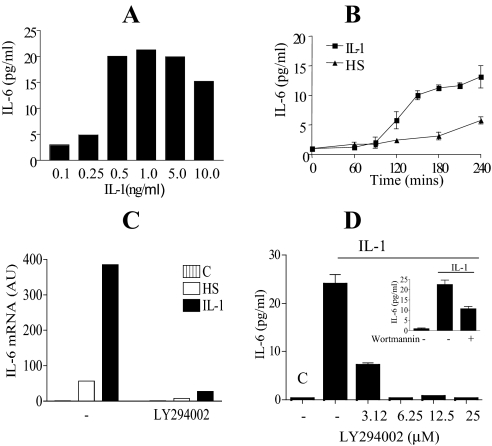
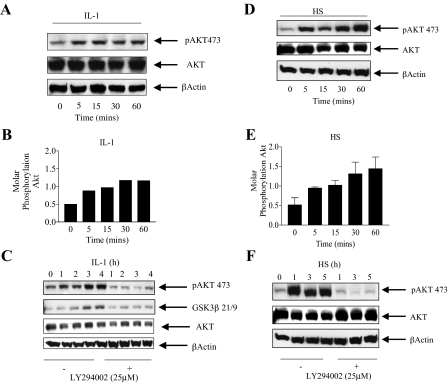
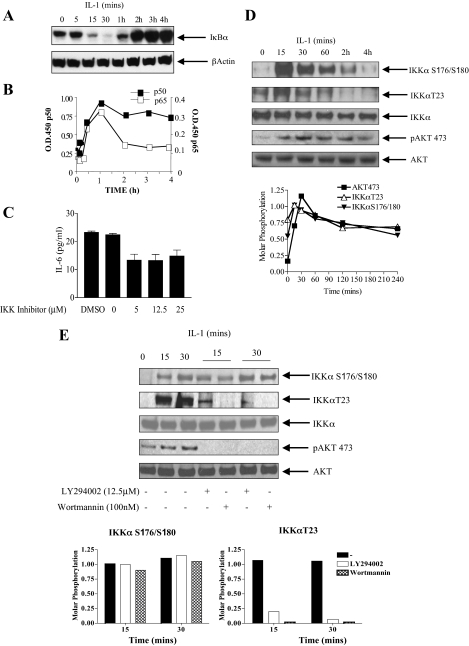
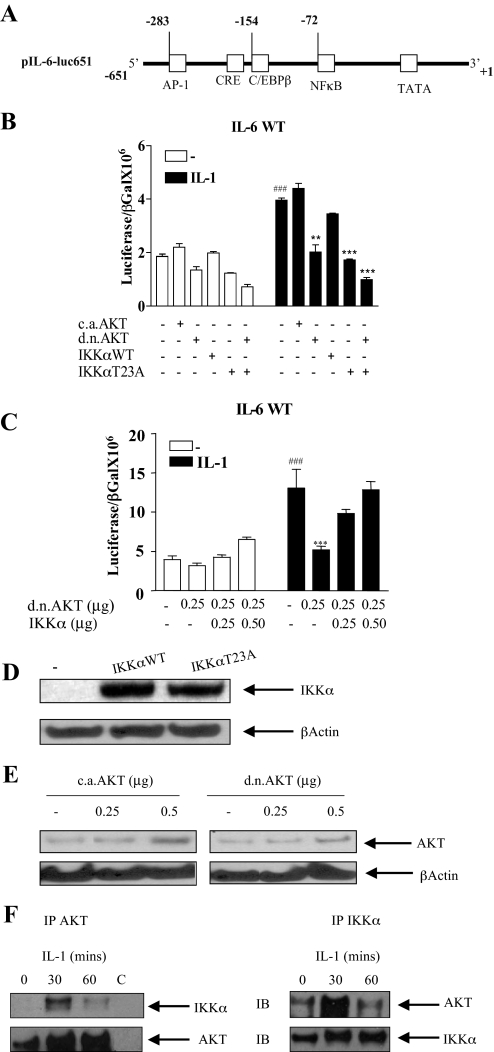
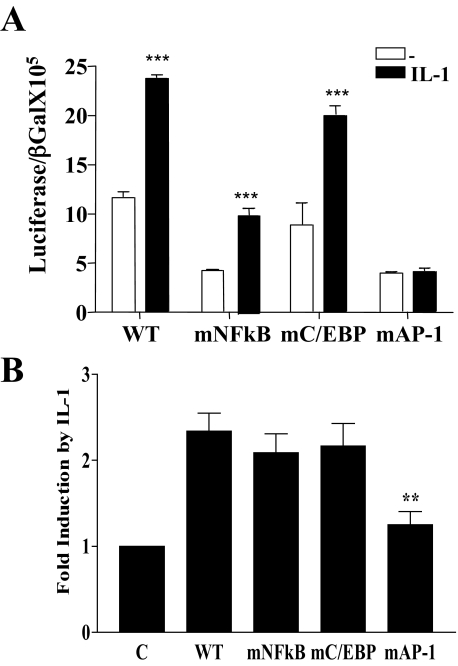
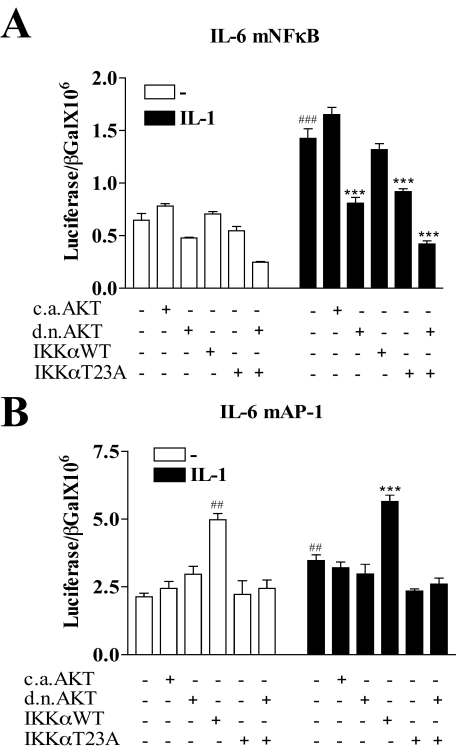
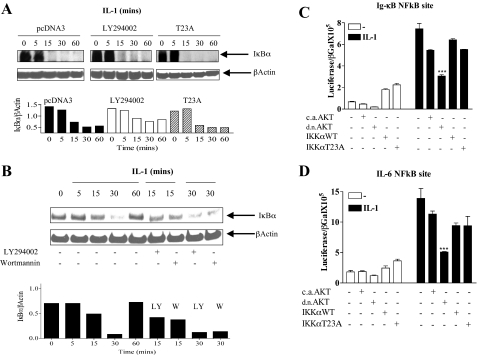
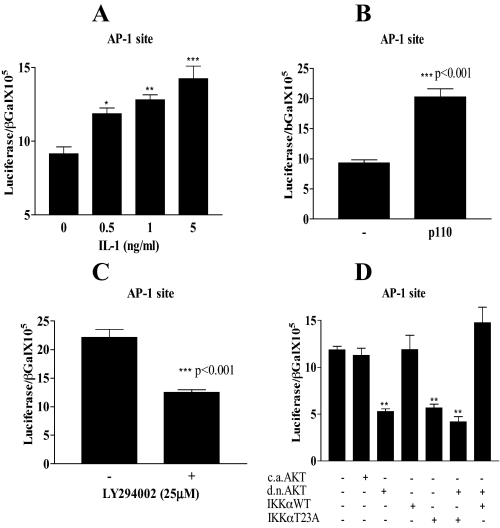
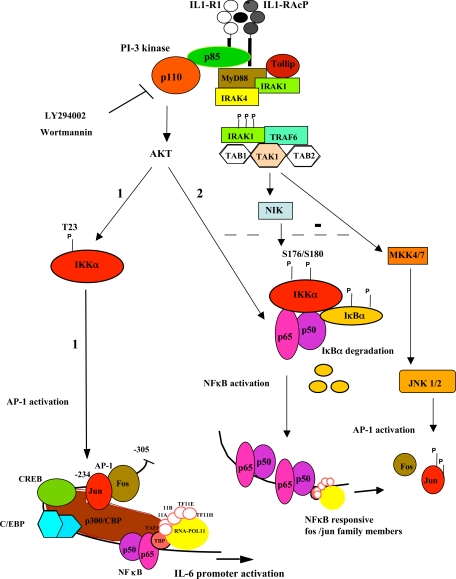
Here we describe a novel role for the phosphatidylinositol 3-kinase/AKT pathway in mediating induction of interleukin-6 (IL-6) in response to IL-1. Pharmacological inhibition of phosphatidylinositol 3-kinase (PI3K) inhibited IL-6 mRNA and protein production. Overexpression of either dominant-negative AKT or IkappaB kinase alpha mutant, IKKalphaT23A, containing a mutation in a functional AKT phosphorylation site, shown previously to be important for NFkappaB activation, completely abrogated IL-6 promoter activation in response to IL-1. However, mutation of the consensus NFkappaB site on the IL-6 promoter did not abrogate promoter activation by IL-1 in contrast to the AP-1 site mutation. IL-1 induces phosphorylation of IKKalpha on the NFkappaB inducing kinase (NIK) phosphorylation sites Ser(176)/Ser(180) and on the Thr(23) site, and although phosphorylation of IKKalphaT23 is inhibited both by LY294002 and wortmannin, phosphorylation of Ser(176)/Ser(180) is not. Neither inhibition of PI 3-kinase/AKT nor IKKalphaT23A overexpression affected IkappaBalpha degradation in response to IL-1. Only partial inhibition by dominant-negative AKT and no inhibitory effect of IKKalphaT23A was observed on an IL-6 promoter-specific NFkappaB site in contrast to significant inhibitory effects on the AP-1 site. Taken together, we have discovered a novel PI 3-kinase/AKT-dependent pathway in response to IL-1, encompassing PI 3-kinase/AKT/IKKalphaT23 upstream of AP-1. This novel pathway is a parallel pathway to the PI 3-kinase/AKT upstream of NFkappaB and both are involved in IL-6 gene transcription in response to IL-1.
Figures
References
-
- Hirano, T. (1998) Int. Rev. Immunol. 16 249–284 - PubMed
-
- Nishimoto, N., and Kishimoto, T. (2006) Nat. Clin. Pract. Rheumatol. 2 619–626 - PubMed
-
- Naugler, W. E., Sakurai, T., Kim, S., Maeda, S., Kim, K., Elsharkawy, A. M., and Karin, M. (2007) Science 317 121–124 - PubMed
-
- Rakoff-Nahoum, S., and Medzhitov, R. (2007) Science 317 124–127 - PubMed
-
- Greten, F. R., Eckmann, L., Greten, T. F., Park, J. M., Li, Z. W., Egan, L. J., Kagnoff, M. F., and Karin, M. (2004) Cell 118 285–296 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
