

Viral control of mitochondrial apoptosis
- PMID: 18516228
- PMCID: PMC2376094
- DOI: 10.1371/journal.ppat.1000018
Viral control of mitochondrial apoptosis
Abstract
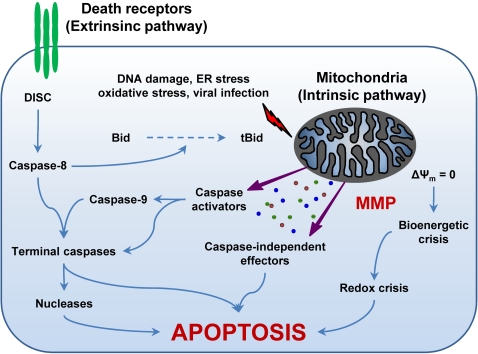
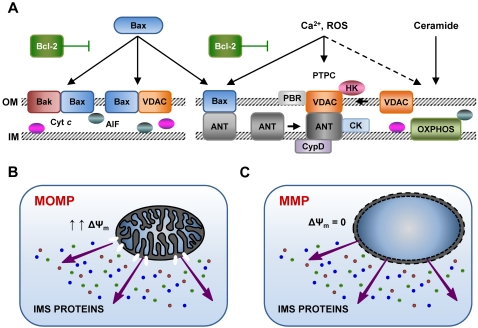
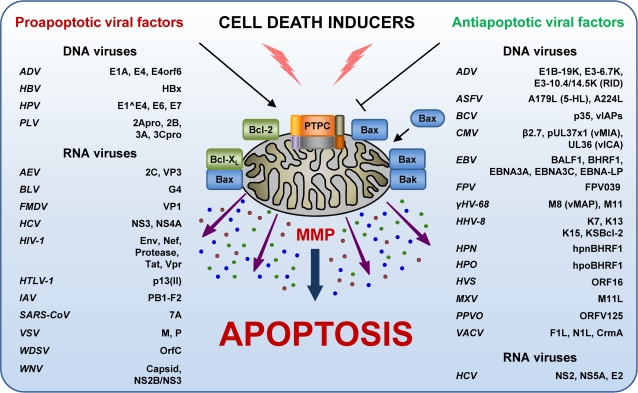
Throughout the process of pathogen-host co-evolution, viruses have developed a battery of distinct strategies to overcome biochemical and immunological defenses of the host. Thus, viruses have acquired the capacity to subvert host cell apoptosis, control inflammatory responses, and evade immune reactions. Since the elimination of infected cells via programmed cell death is one of the most ancestral defense mechanisms against infection, disabling host cell apoptosis might represent an almost obligate step in the viral life cycle. Conversely, viruses may take advantage of stimulating apoptosis, either to kill uninfected cells from the immune system, or to induce the breakdown of infected cells, thereby favoring viral dissemination. Several viral polypeptides are homologs of host-derived apoptosis-regulatory proteins, such as members of the Bcl-2 family. Moreover, viral factors with no homology to host proteins specifically target key components of the apoptotic machinery. Here, we summarize the current knowledge on the viral modulation of mitochondrial apoptosis, by focusing in particular on the mechanisms by which viral proteins control the host cell death apparatus.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. - PubMed
-
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. - PubMed
-
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, et al. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. - PubMed
-
- De Maria R, Lenti L, Malisan F, d'Agostino F, Tomassini B, et al. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science. 1997;277:1652–1655. - PubMed
-
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–786. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
