

The evolutionary genetics and emergence of avian influenza viruses in wild birds
- PMID: 18516303
- PMCID: PMC2387073
- DOI: 10.1371/journal.ppat.1000076
The evolutionary genetics and emergence of avian influenza viruses in wild birds
Abstract
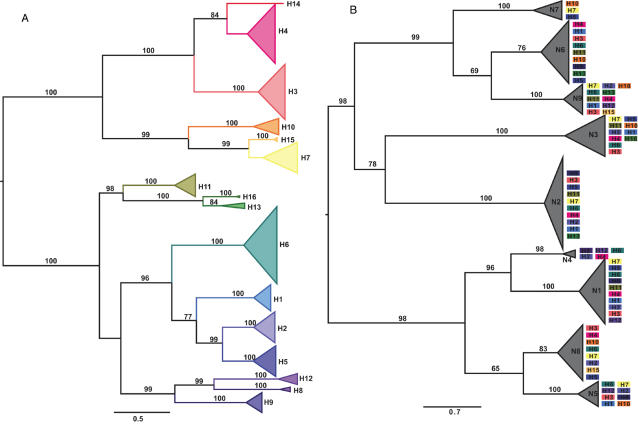
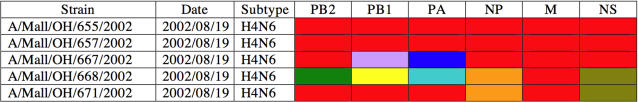
We surveyed the genetic diversity among avian influenza virus (AIV) in wild birds, comprising 167 complete viral genomes from 14 bird species sampled in four locations across the United States. These isolates represented 29 type A influenza virus hemagglutinin (HA) and neuraminidase (NA) subtype combinations, with up to 26% of isolates showing evidence of mixed subtype infection. Through a phylogenetic analysis of the largest data set of AIV genomes compiled to date, we were able to document a remarkably high rate of genome reassortment, with no clear pattern of gene segment association and occasional inter-hemisphere gene segment migration and reassortment. From this, we propose that AIV in wild birds forms transient "genome constellations," continually reshuffled by reassortment, in contrast to the spread of a limited number of stable genome constellations that characterizes the evolution of mammalian-adapted influenza A viruses.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Easterday BC, Trainer DO, Tumova B, Pereira HG. Evidence of infection with influenza viruses in migratory waterfowl. Nature. 1968;219:523–524. - PubMed
-
- Slemons RD, Johnson DC, Osborn JS, Hayes F. Type-A influenza viruses isolated from wild free-flying ducks in California. Avian Dis. 1974;18:119–124. - PubMed
-
- Krauss S, Walker D, Pryor SP, Niles L, Chenghong L, et al. Influenza A viruses of migrating wild aquatic birds in North America. Vector Borne Zoonotic Dis. 2004;4:177–189. - PubMed
-
- Spackman E, Stallknecht DE, Slemons RD, Winker K, Suarez DL, et al. Phylogenetic analyses of type A influenza genes in natural reservoir species in North America reveals genetic variation. Virus Res. 2005;114:89–100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
