

AlkB homologue 2-mediated repair of ethenoadenine lesions in mammalian DNA
- PMID: 18519673
- PMCID: PMC4725713
- DOI: 10.1158/0008-5472.CAN-08-0796
AlkB homologue 2-mediated repair of ethenoadenine lesions in mammalian DNA
Abstract
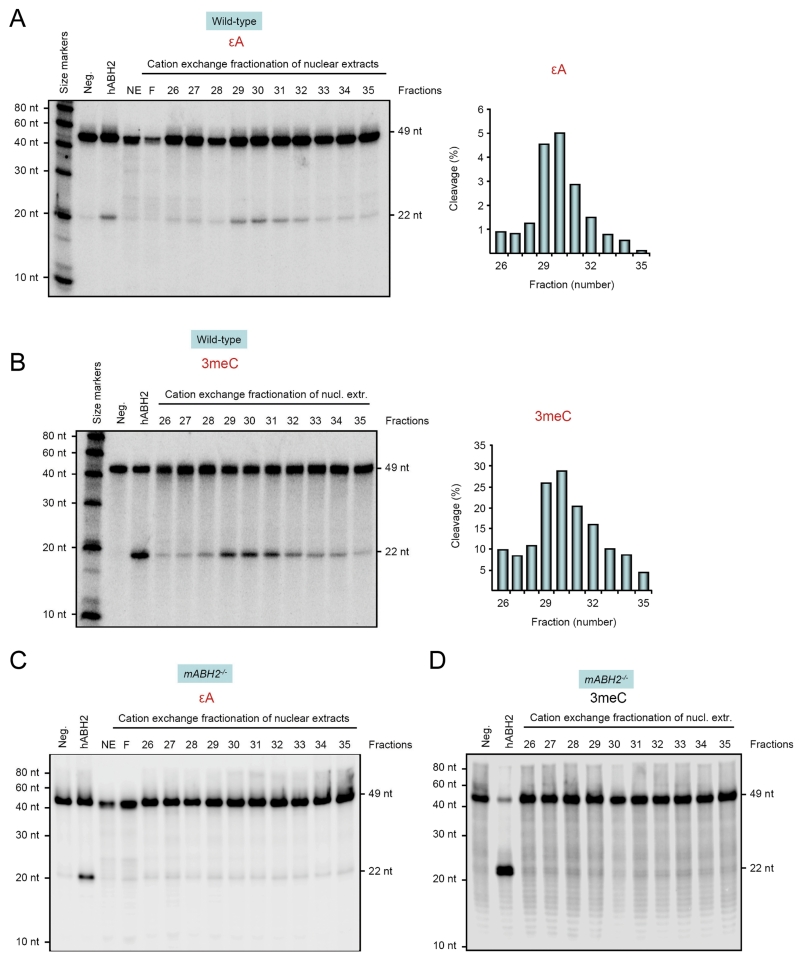
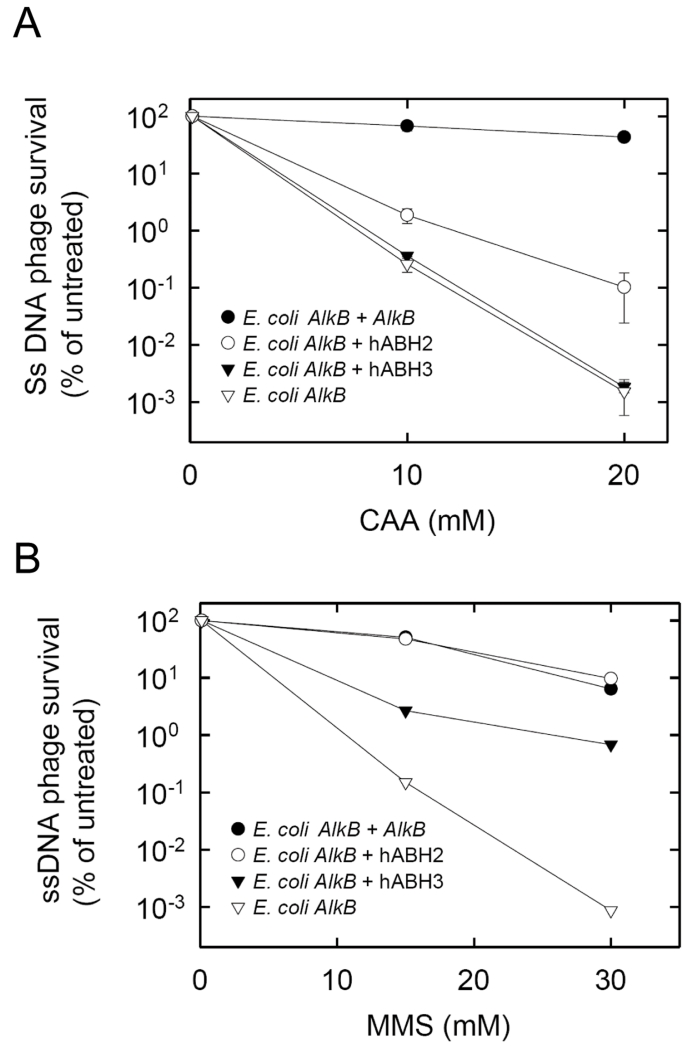
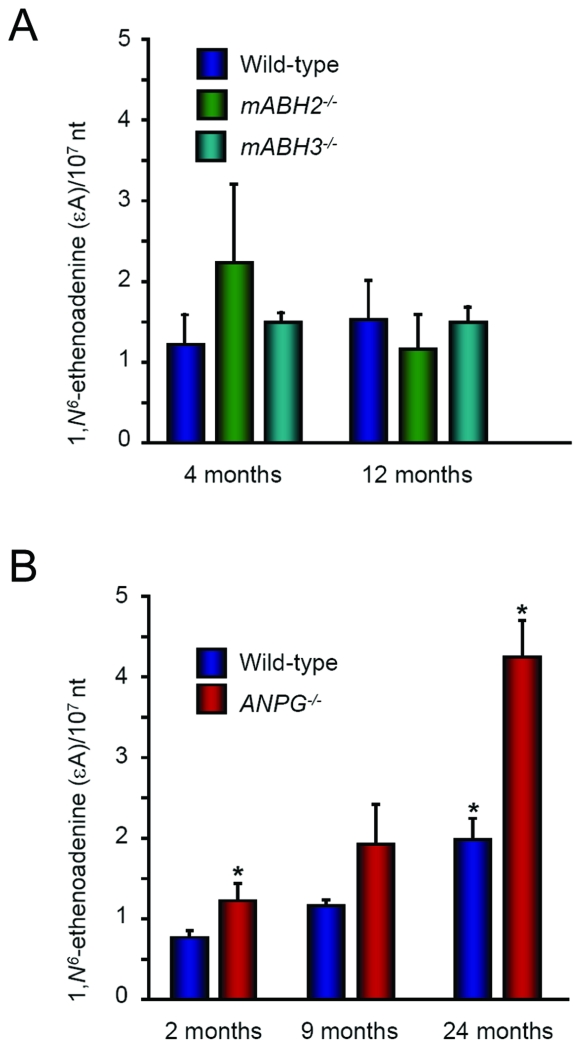
Endogenous formation of the mutagenic DNA adduct 1,N(6)-ethenoadenine (epsilon A) originates from lipid peroxidation. Elevated levels of epsilon A in cancer-prone tissues suggest a role for this adduct in the development of some cancers. The base excision repair pathway has been considered the principal repair system for epsilon A lesions until recently, when it was shown that the Escherichia coli AlkB dioxygenase could directly reverse the damage. We report here kinetic analysis of the recombinant human AlkB homologue 2 (hABH2), which is able to repair epsilon A lesions in DNA. Furthermore, cation exchange chromatography of nuclear extracts from wild-type and mABH2(-/-) mice indicates that mABH2 is the principal dioxygenase for epsilon A repair in vivo. This is further substantiated by experiments showing that hABH2, but not hABH3, is able to complement the E. coli alkB mutant with respect to its defective repair of etheno adducts. We conclude that ABH2 is active in the direct reversal of epsilon A lesions, and that ABH2, together with the alkyl-N-adenine-DNA glycosylase, which is the most effective enzyme for the repair of epsilon A, comprise the cellular defense against epsilon A lesions.
Figures



Similar articles
-
The role of AlkB protein in repair of 1,N⁶-ethenoadenine in Escherichia coli cells.Mutagenesis. 2011 May;26(3):401-6. doi: 10.1093/mutage/geq107. Epub 2010 Dec 30. Mutagenesis. 2011. PMID: 21193516
-
AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo.Nat Struct Mol Biol. 2005 Oct;12(10):855-60. doi: 10.1038/nsmb996. Epub 2005 Oct 2. Nat Struct Mol Biol. 2005. PMID: 16200073
-
Insights into the Direct Oxidative Repair of Etheno Lesions: MD and QM/MM Study on the Substrate Scope of ALKBH2 and AlkB.DNA Repair (Amst). 2020 Dec;96:102944. doi: 10.1016/j.dnarep.2020.102944. Epub 2020 Sep 9. DNA Repair (Amst). 2020. PMID: 33161373 Free PMC article.
-
Enzymology of repair of etheno-adducts.Mutat Res. 2003 Oct 29;531(1-2):219-29. doi: 10.1016/j.mrfmmm.2003.07.008. Mutat Res. 2003. PMID: 14637257 Review.
-
DNA repair: bioinformatics helps reverse methylation damage.Curr Biol. 2002 Dec 23;12(24):R846-8. doi: 10.1016/s0960-9822(02)01350-7. Curr Biol. 2002. PMID: 12498704 Review.
Cited by
-
Inflammation-induced DNA damage, mutations and cancer.DNA Repair (Amst). 2019 Nov;83:102673. doi: 10.1016/j.dnarep.2019.102673. Epub 2019 Jul 25. DNA Repair (Amst). 2019. PMID: 31387777 Free PMC article. Review.
-
Repair of DNA Alkylation Damage by the Escherichia coli Adaptive Response Protein AlkB as Studied by ESI-TOF Mass Spectrometry.J Nucleic Acids. 2010 Oct 27;2010:369434. doi: 10.4061/2010/369434. J Nucleic Acids. 2010. PMID: 21048928 Free PMC article.
-
Repair of endogenous DNA base lesions modulate lifespan in mice.DNA Repair (Amst). 2014 Sep;21:78-86. doi: 10.1016/j.dnarep.2014.05.012. Epub 2014 Jun 30. DNA Repair (Amst). 2014. PMID: 24994062 Free PMC article.
-
Computational investigations of selected enzymes from two iron and α-ketoglutarate-dependent families.Phys Chem Chem Phys. 2021 Oct 13;23(39):22227-22240. doi: 10.1039/d1cp03800a. Phys Chem Chem Phys. 2021. PMID: 34586107 Free PMC article. Review.
-
3,N4-Etheno-5-methylcytosine blocks TET1-3 oxidation but is repaired by ALKBH2, 3 and FTO.Nucleic Acids Res. 2024 Nov 11;52(20):12378-12389. doi: 10.1093/nar/gkae818. Nucleic Acids Res. 2024. PMID: 39315710 Free PMC article.
References
-
- Nair J, Barbin A, Guichard Y, Bartsch H. 1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytine in liver DNA from humans and untreated rodents detected by immunoaffinity/32P-postlabeling. Carcinogenesis. 1995;16:613–7. - PubMed
-
- Laib RJ. The role of cyclic base adducts in vinyl-chloride-induced carcinogenesis: studies on nucleic acid alkylation in vivo. IARC Sci Publ. 1986;70:101–8. - PubMed
-
- Levine RL, Yang IY, Hossain M, Pandya GA, Grollman AP, Moriya M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 2000;60:4098–104. - PubMed
-
- Pandya GA, Moriya M. 1,N6-ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry. 1996;35:11487–92. - PubMed
-
- Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg. 2006;391:499–510. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
