

Redox regulation of cell survival
- PMID: 18522489
- PMCID: PMC2932530
- DOI: 10.1089/ars.2007.1957
Redox regulation of cell survival
Abstract
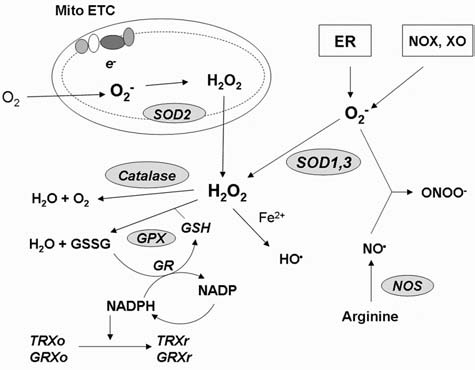
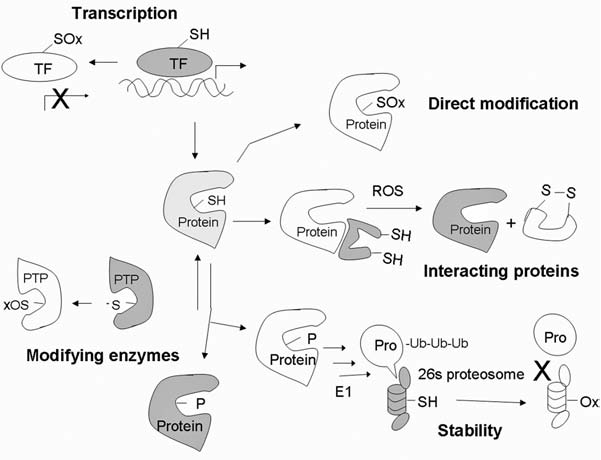
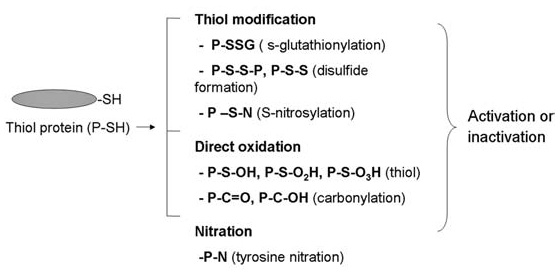
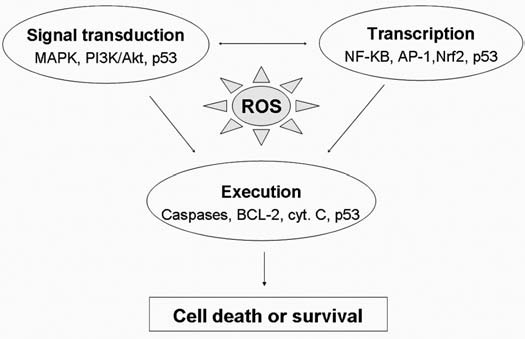
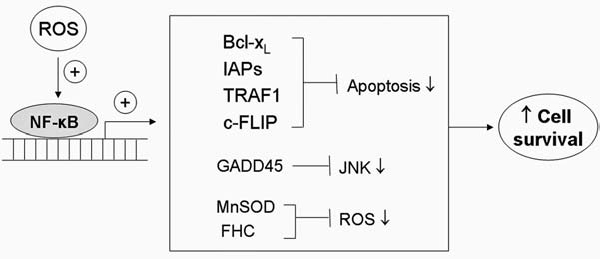
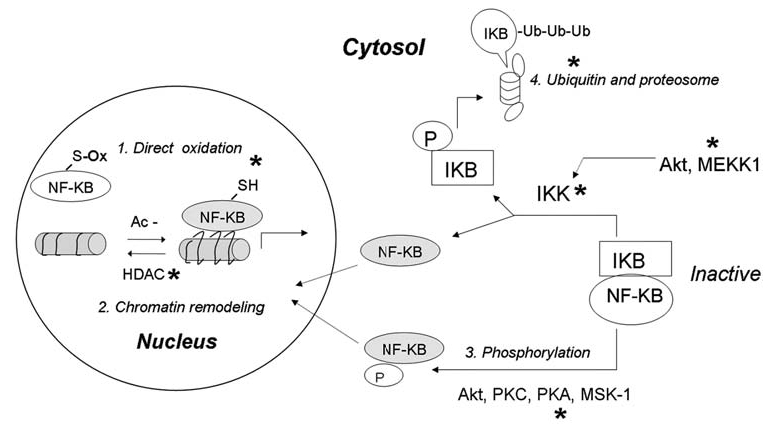
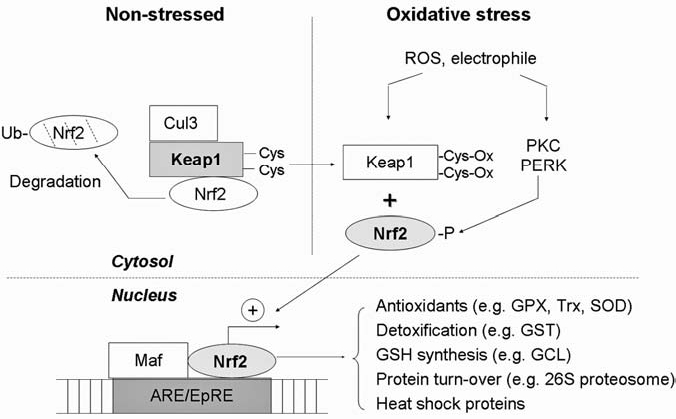
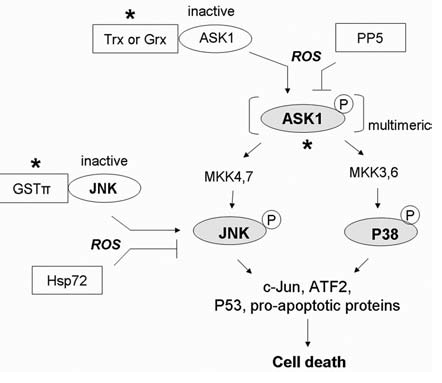
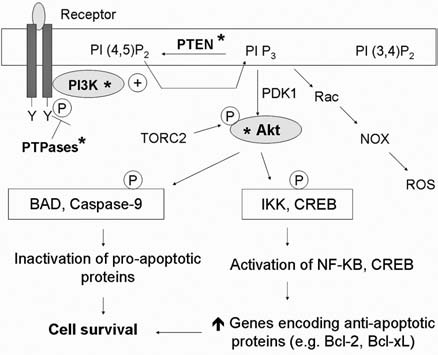
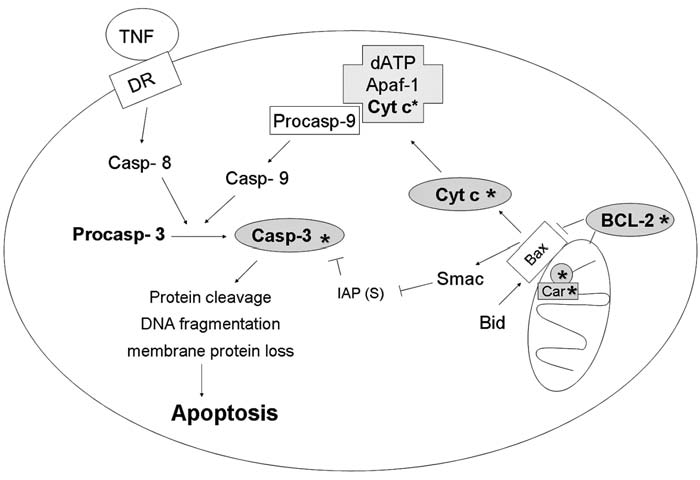
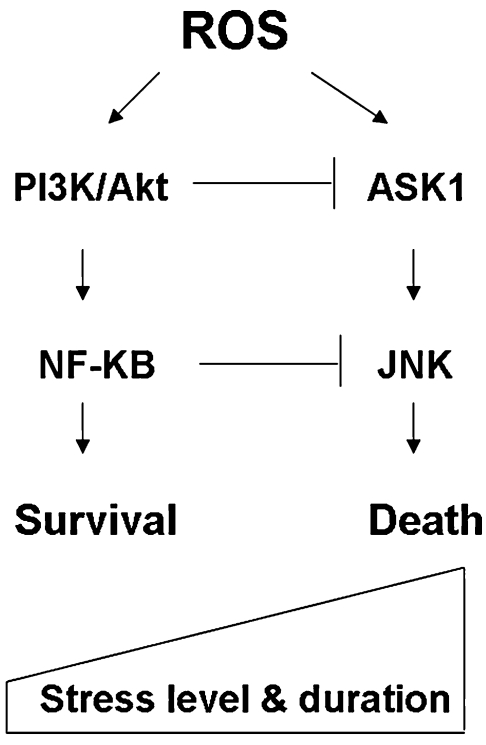
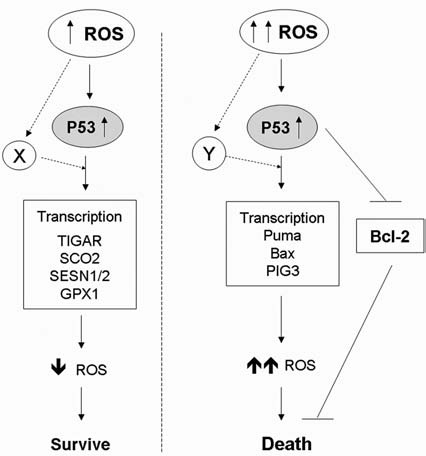
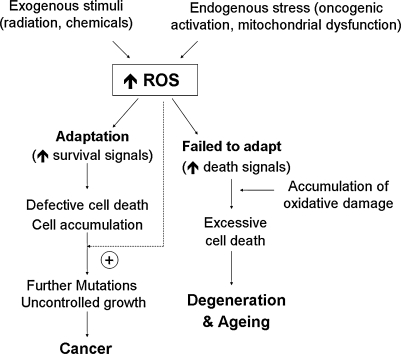
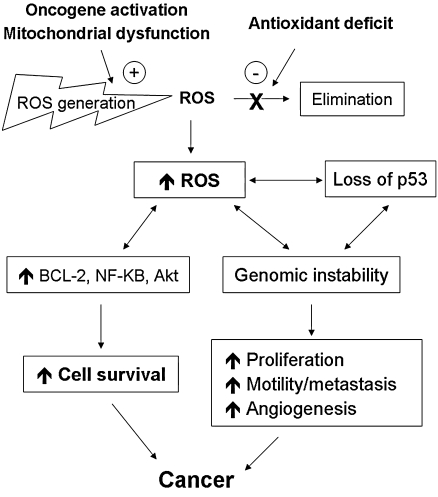
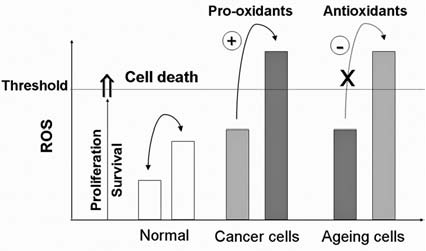
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) play important roles in regulation of cell survival. In general, moderate levels of ROS/RNS may function as signals to promote cell proliferation and survival, whereas severe increase of ROS/RNS can induce cell death. Under physiologic conditions, the balance between generation and elimination of ROS/RNS maintains the proper function of redox-sensitive signaling proteins. Normally, the redox homeostasis ensures that the cells respond properly to endogenous and exogenous stimuli. However, when the redox homeostasis is disturbed, oxidative stress may lead to aberrant cell death and contribute to disease development. This review focuses on the roles of key transcription factors, signal-transduction pathways, and cell-death regulators in affecting cell survival, and how the redox systems regulate the functions of these molecules. The current understanding of how disturbance in redox homeostasis may affect cell death and contribute to the development of diseases such as cancer and degenerative disorders is reviewed. We also discuss how the basic knowledge on redox regulation of cell survival can be used to develop strategies for the treatment or prevention of those diseases.
Figures
References
-
- Alexandre J. Nicco C. Chereau C. Laurent A. Weill B. Goldwasser F. Batteux F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006;98:236–244. - PubMed
-
- Angel P. Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. - PubMed
-
- O'Brian CA. Chu F. Review: post-translational disulfide modifications in cell signaling: role of inter-protein, intra-protein, S-glutathionyl, and S-cysteaminyl disulfide modifications in signal transmission. Free Radic Res. 2005;39:471–480. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
