

Marine actinomycetes: a new source of compounds against the human malaria parasite
- PMID: 18523554
- PMCID: PMC2391291
- DOI: 10.1371/journal.pone.0002335
Marine actinomycetes: a new source of compounds against the human malaria parasite
Abstract
Background: Malaria continues to be a devastating parasitic disease that causes the death of 2 million individuals annually. The increase in multi-drug resistance together with the absence of an efficient vaccine hastens the need for speedy and comprehensive antimalarial drug discovery and development. Throughout history, traditional herbal remedies or natural products have been a reliable source of antimalarial agents, e.g. quinine and artemisinin. Today, one emerging source of small molecule drug leads is the world's oceans. Included among the source of marine natural products are marine microorganisms such as the recently described actinomycete. Members of the genus Salinispora have yielded a wealth of new secondary metabolites including salinosporamide A, a molecule currently advancing through clinical trials as an anticancer agent. Because of the biological activity of metabolites being isolated from marine microorganisms, our group became interested in exploring the potential efficacy of these compounds against the malaria parasite.
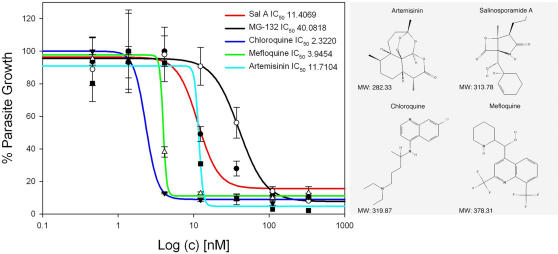
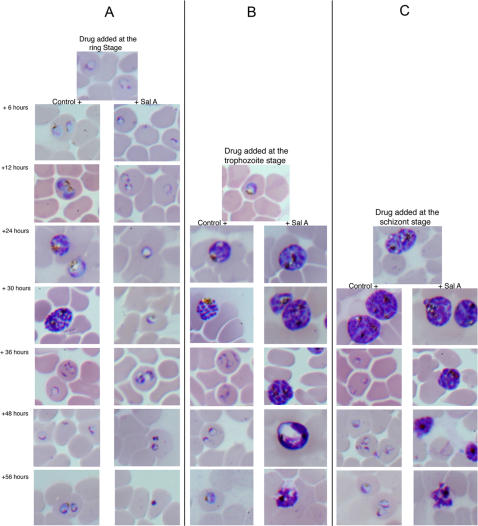
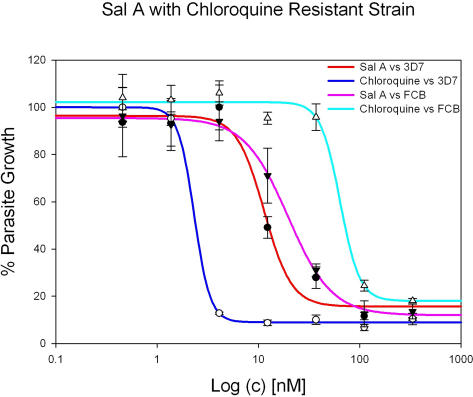
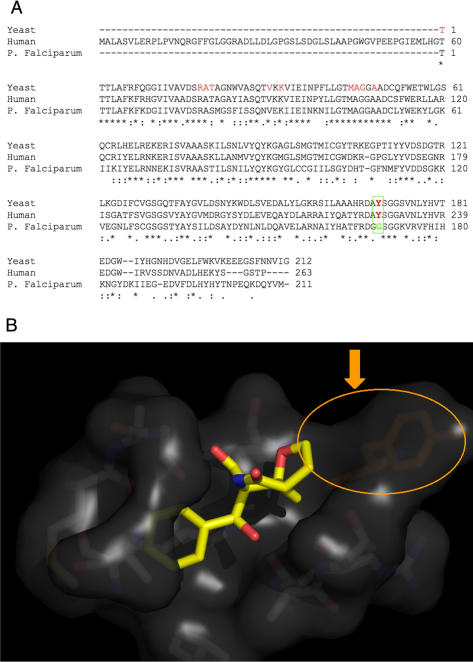
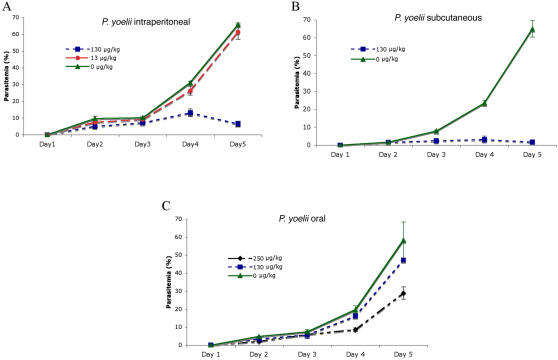
Methods: We screened 80 bacterial crude extracts for their activity against malaria growth. We established that the pure compound, salinosporamide A, produced by the marine actinomycete, Salinispora tropica, shows strong inhibitory activity against the erythrocytic stages of the parasite cycle. Biochemical experiments support the likely inhibition of the parasite 20S proteasome. Crystal structure modeling of salinosporamide A and the parasite catalytic 20S subunit further confirm this hypothesis. Ultimately we showed that salinosporamide A protected mice against deadly malaria infection when administered at an extremely low dosage.
Conclusion: These findings underline the potential of secondary metabolites, derived from marine microorganisms, to inhibit Plasmodium growth. More specifically, we highlight the effect of proteasome inhibitors such as salinosporamide A on in vitro and in vivo parasite development. Salinosporamide A (NPI-0052) now being advanced to phase I trials for the treatment of refractory multiple myeloma will need to be further explored to evaluate the safety profile for its use against malaria.
Conflict of interest statement
Figures

References
-
- Ferrer-Rodriguez I, Perez-Rosado J, Gervais GW, Peters W, Robinson BL, et al. Plasmodium yoelii: identification and partial characterization of an MDR1 gene in an artemisinin-resistant line. J Parasitol. 2004;90:152–160. - PubMed
-
- Jambou R, Legrand E, Niang M, Khim N, Lim P, et al. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet. 2005;366:1960–1963. - PubMed
-
- Alker AP, Lim P, Sem R, Shah NK, Yi P, et al. Pfmdr1 and in vivo resistance to artesunate-mefloquine in falciparum malaria on the Cambodian-Thai border. Am J Trop Med Hyg. 2007;76:641–647. - PubMed
-
- Wan Y, Hur W, Cho CY, Liu Y, Adrian FJ, et al. Synthesis and target identification of hymenialdisine analogs. Chem Biol. 2004;11:247–259. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
